Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ester Blanco and Version 2 by Rita Xu.
Immunotherapies based on immune checkpoint blockade have shown remarkable clinical outcomes and durable responses in patients with many tumor types. These therapies lack efficacy in most cancer patients, even causing severe adverse events in a small subset of patients, such as inflammatory disorders and hyper-progressive disease. To diminish the risk of developing serious toxicities, intratumor delivery of monoclonal antibodies could be a solution. Encouraging results have been shown in both preclinical and clinical studies.
- monoclonal antibodies (mAbs)
- intratumoral therapy
- viral therapy
1. Therapeutic Antibodies: Beyond Conventional Monoclonal Antibodies (mAbs)
In 1988, Greg Winter and his team pioneered the technique to humanize monoclonal antibodies, and since then the field of personalized therapy using mAbs has been successfully developed for the treatment of various cancers [1]. To date, approximately more than 100 mAbs have been approved by the US Food and Drug Administration (FDA) for the treatment of cancer [2].
Monoclonal antibodies are engineered to specifically bind target antigens with high affinity. These antibodies then have the potential to induce complement-dependent cytotoxicity and promote innate and adaptive immune responses depending on their target specificities [3]. MAbs are large immunoglobulin molecules, usually in the range of about 150 kDa. The standard mAb molecule is composed of four polypeptide chains, forming a Y-shaped macromolecule complex. More specifically, mAbs are made of two identical heavy chains and two identical light chains connected by interchain disulphide bonds and non-covalent interactions. The light chain and two N-terminal domains of the heavy chain comprise the antigen-binding fragment (Fab). The variable domains of the heavy (VH) and light (VL) chains form the antigen recognition domain by adopting a structure with three hypervariable loops in each domain that comprise the complementary-determining regions (CDRs 1, 2, and 3), flanked by four conserved framework regions (FRs 1, 2, 3, and 4) (Figure 1A). The immune-mediated functions, such as antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC), are conferred by the C-terminal halves of the heavy chains, which comprise the crystallizable fragment (Fc). The conserved domains of the heavy chains differ between distinct antibody classes (isotypes). From these, the immunoglobulin G (IgG) isotypes are the main ones in human plasma. Hence, IgG-based monoclonal antibodies (mAbs) have become a dominant class of biotherapeutics in recent decades [4].
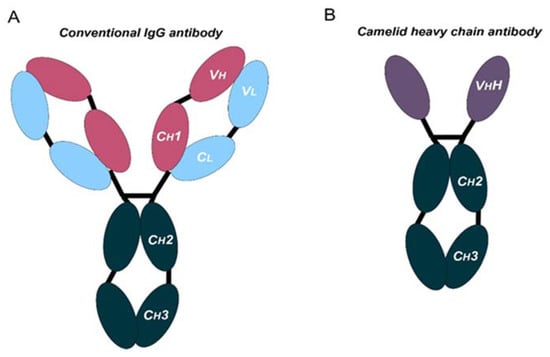
Figure 1. Schematic representation of the structure of conventional and camelid heavy-chain antibodies. (A) Structure of a conventional IgG antibody, composed by two heavy and two light chains. Conserved and variable heavy-chain domains (Ch, Vh) and light domains (Cl, Vl) are indicated. (B) Structure of a camelid heavy-chain antibody, composed by two heavy chains.
In the last decade, mAbs that block immune checkpoint molecules have shown remarkable clinical outcomes and durable responses in treated cancer patients. These mAbs have been engineered to reactivate antitumor immunity by blockade of T cell inhibitory molecules (immune checkpoints, IC), such as CTLA-4, PD-1, PD-L1, and LAG-3, which are the most studied IC. These molecules are expressed in different immune or non-immune cells. PD-L1 can be expressed by cancer cells and myeloid cells [5]. In addition, antigen-presenting cells (APC) express CTLA-4, and T cells and NK cells express PD-1, LAG-3, and other molecules, such as TIM-3 and TIGIT [6].
Antibodies targeting these latter targets are widely used for the management of cancer [7][8][9][10][11][12][13][7,8,9,10,11,12,13]. These therapies rely on systemic administration of these mAbs every three to four weeks. Moreover, these mAbs frequently present poor tissue penetration, intrinsic immunogenicity, and high production costs. All these issues suggest that there is still room for improvement. In fact, the large and complex structure of these mAbs causes several disadvantages: (1) high production cost due to their complexity and posttranslational modifications, (2) limited physicochemical stability, (3) systemic administration, which may cause off-target immune-related toxicities, (4) low penetration in solid tumors, and (5) poor penetration in the brain [14][15][14,15]. In certain cases, the production and use of full-length antibodies may be problematic, as mentioned above. Recent advances in antibody engineering have facilitated the production of a collection of antibody variants for the use in cancer, including other antibody formats (e.g., antibody fragments, bispecific antibodies (BsAbs), and non-IgG scaffold proteins) and antibody derivatives (e.g., antibody–drug conjugates (ADCs) and immunocytokines) [16]. Right now, the field of antibodies is revolving around the engineering of the antigen-binding region from heavy single-chain antibodies (HCAbs), such as those from camelids (dromedaries, camels, llamas, alpacas, guanacos, and vicuñas), coined with the term “nanobodies” [17]. Compared to conventional mAbs, HCAbs are homodimers made of two identical heavy-chain molecules, lacking both the light chain and the constant domain 1 (CH1) of the heavy chain. These antibodies possess a significantly smaller molecular mass, of approximately 95 kDa (Figure 1B). The variable antigen-binding domain of HCAbs (called VHH) retains full antigen-binding potential despite lacking the light chain. Indeed, the VHH domain is the smallest naturally occurring antigen-binding fragment [18].
The discovery of HCAbs has spurred the development of nanobody production platforms due to their inherent properties, which make them very attractive tools for cancer treatment: (1) nanoscale dimensions enable deeper tumor penetration, (2) certain nanobodies can cross the blood–brain barrier (BBB) [19], and (3) high affinity and specificity for their targets with low off-target accumulation. Structurally, nanobodies are similar to the VH domain of conventional antibodies, with four FRs and three CDRs. Besides, nanobodies exhibit high homology with the VH domain of human family III immunoglobulins. In addition, nanobodies present higher hydrophilicity, with increased solubility and enhanced physicochemical stability compared to the variable fragments from conventional antibodies. Moreover, the CDR3 motif tends to be longer, being the main region implicated in antigen binding. The antigen-binding region is made by CDR3 and CDR2, together with some FR residues. This extended CDR3 loop allows binding to small cavities or concave epitopes (mainly conformational epitopes), such as catalytic sites of enzymes. In contrast, conventional antibodies are better at recognizing small chemical groups (haptens), peptides, or flat epitopes on proteins [20][21][20,21]. Currently, several nanobody-based therapeutics are under clinical trials for the treatment of a variety of diseases, including cancer, autoimmune diseases, and viral infections [22].
2. Routes of mAbs Administration
The success of immune checkpoint blockade immunotherapies has demonstrated significant advances in the treatment of many cancers. However, current immunotherapies fail in most cancer patients. There are many reasons for the failure of immunotherapies, which include poor immunogenicity characterized by reduced tumor infiltration with immune cells, and systemic immune dysfunctionality [9][23][24][25][9,37,38,39]. The lack of penetration of mAbs within the tumor environment adds up to the poor immunogenicity of many cancer types. These problems contribute to the failure of ICB therapies in many cancer patients. The systemic parenteral use of therapeutic mAbs has unequivocal advantages, allowing simplicity of administration and predictable serum pharmacokinetics. However, this mode of delivery presents limitations and disadvantages, which include poor penetration into solid tumors and systemic toxicities caused by off-target effects, with systemic inflammation and autoimmune or autoimmune-like reactions [26][27][40,41]. Indeed, serious immune-related adverse events (irAEs) are associated with recurrent systemic administration of ICB antibodies [28][29][30][42,43,44]. These constraints could be overcome, or at least reduced, by enhancing mAbs availability within the tumor microenvironment (TME). One way to achieve this would be through intratumor delivery of mAbs and locoregional delivery. Several routes have been tested for mAbs delivery, for example subcutaneous (SC), intramuscular (IM), oral, and intratumor (I.T) administration. SC injection would be the most convenient for patients. Thus, various mAbs have emerged, designed for subcutaneous administration [31][32][45,46]. This administration route would be suited for self-administration by the patient, although so far, the accuracy and efficacy of mAbs delivered in such a way is hard to predict. Indeed, this is a major issue for human therapy. In addition, the mAb formulation itself for SC delivery is still a major challenge for drug development [33][47]. The same arguments can be applied for muscular delivery. Nevertheless, the clinical application of these type of drugs for these administration routes is hampered by their complex structure. Oral administration of mAbs has been discarded because of inefficient transport through tissue barriers such as the intestinal mucosa, while they are quickly degraded by proteases in the gastrointestinal tract. In contrast to other strategies, intratumor delivery can increase the therapeutic index of mAbs by restricting them to within the tumor environment, with a reduced risk for off-target toxicities. In addition, repeated intratumor injections can be administered, leading to much higher local bioactive drug concentrations [34][35][48,49]. Despite the challenges associated with intratumor delivery, it has substantial potential to improve immunotherapies.3. Intratumor mAbs Delivery in Solid Tumors
Intratumor administration using image-guided injection is achievable for most organs [34][48]. Following administration, the therapeutic agents first diffuse throughout the injected area, thereby achieving a very high local concentration. Overtime, drugs will dissipate into systemic circulation. In fact, this gradual absorption into the blood can have pharmacokinetic advantages that will permit higher local doses with better tolerability, as shown for other protein-based drugs [36][50] (NCT02304393). Importantly, intratumor delivery allows the immediate access to tumor-draining lymph nodes and other lymphoid structures within the tumor tissue, amplifying the immune response [37][38][39][51,52,53]. Importantly, intratumor administration of mAbs and other drugs shows an abscopal effect in distal metastases, indicating that this administration route can have systemic activities [40][41][54,55]. Injections in multiple tumor lesions within the same patient can also enhance polyclonal responses, despite the high variability of cancer cells in metastases [42][56]. In addition, several studies have evaluated intratumoral mAbs and short peptides against receptors expressed on the blood–brain barrier (BBB) delivery to achieve brain targeting [43][44][57,58]. Several drug combinations have been evaluated with success in preclinical studies, for example combinations of antibodies targeting CTLA4, OX40, PD-1, and CD137 [45][59]. Some formulations also allow a slow intratumor release of mAbs, which leads to a prolonged and improved therapeutic index [46][60]. There are two main vector strategies for optimized intratumor mAbs administration: non-viral- and viral-based vectors. Both strategies present intrinsic advantages and disadvantages (Figure 2). The efficiency of modifying host cells with mAb-encoding nucleic acids is better with viral vectors, but a major drawback of viral vectors is their immunogenicity and potential cytotoxicity [47][48][61,62]. Replicative oncolytic viruses are also emerging as promising anti-cancer treatments, but in this case, these can be regarded as an in-situ treatment that releases antigens and damage-associated molecules, rather than virus-based vectors for drug delivery. On the other hand, non-viral vectors possess better safety profiles but less efficient capacities to modify target cells in vivo [49][50][63,64].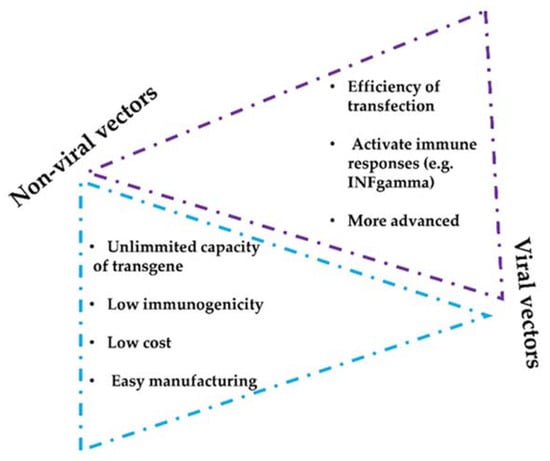
Figure 2. Considerations of non-viral and viral vectors for mAbs intratumor delivery systems. The figure shows the main advantages and limitations that distinguish viral and non-viral strategies.