Ionotropic receptors for (
S)-glutamate (iGluR), the main excitatory neurotransmitter in the mammalian central nervous system (CNS), are associated with Na
+, K
+ and Ca
2+ ion channels and are responsible for the majority of rapid excitatory neurotransmission in the CNS. The iGluR family comprises four functional classes, including N-methyl-D-aspartate receptors (NMDAR), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR), kainate receptors (KAR) and delta receptors. As with other iGluRs, kainate receptors function as tetramers (dimers of dimers), which are composed of individual subunits GluK1 to GluK5. Based on the affinity of kainic acid, the KAR subfamily is divided into low-affinity subunits GluK1–3, which can form functional homomeric or heteromeric ion channels, and high-affinity subunits GluK4–5 that form functional receptors only in combination with subunits GluK1–3
[1][2][3]. KARs are highly expressed in the CNS, particularly in the hippocampal formation, lateral amygdala, dorsal root ganglia, bipolar cells of the retina, cerebral cortex, and cerebellum. GluK2/GluK5 receptors represent a major population of KARs in the brain, while the expression of other subunits varies according to location, cell types, synapses, and developmental stages; for example, GluK3 is expressed primarily in the dentate gyrus in the hippocampus as well as neocortex regions
[1][4][5][6][7][8].
2. Chemistry
The synthesis of the target quinoxaline-2,3-diones (
5–29) was achieved through the synthetic route presented in Scheme 1, following a modified procedure described by Pallesen for compound
1 [32]. Commercially available 1-fluoro-4-iodo-2-nitrobenzene
2 was transformed into
3 through an S
NAr reaction using benzohydrazide. The next step proceeded as a one-pot procedure through acylation, reduction of the nitro group, and spontaneous ring closure. The Sonogashira cross-coupling reaction allowed the introduction of a triple C-C bond moiety in place of iodine and the obtaining of the target structures (
5–29).
Scheme 1. Synthesis of target compounds 5–29: (a) benzohydrazide, K2CO3, DMSO, rt, 12 h; (b) ethyl chlorooxoacetate, triethylamine, THF, rt, 30 min; (c) iron powder, acetic acid, reflux, 30 min; (d) appropriate arylethynyl reagent, PdCl2(PPh3)2, CuI, triethylamine, DMF.
3. Pharmacological Characterization
3.1. Binding Pharmacology
The final compounds were characterized in binding studies using homomeric recombinant rat iGluRs: GluK1-GluK3, GluK5, and GluA2, expressed in Sf9 insect cell membranes. The in vitro binding data are presented in Table 1.
The observed results suggest that the introduction of the arylethynyl moiety to the quinoxaline-2,3-dione core is strongly related to the effect of preferential binding to GluK3 receptors. Most synthesized quinoxaline-2,3-diones showed measurable affinity exclusively at GluK3 (or both GluK3 and GluK1) homomeric receptors. Among the compounds tested at the receptors GluK2, GluK5 and GluA2, all analogues except 27 and 29 were found to be inactive (Ki > 100 μM, Table 1).
Within the series obtained, compounds 5–13 were designed to explore the available steric space of the GluK3 binding site and contained an additional lipophilic substituent in the arylethynyl moiety. In general, the introduction of lipophilic groups at position 4 of the phenyl ring (6–13) resulted in a drop of GluK3 binding: the more significant, the larger the size of the substituent. Similarly, a decrease in GluK3 affinity was observed for most polar modifications (14–24), which were intended to explore the potential for hydrogen bond formation in this part of the receptor-binding pocket. Only selected substituents at the 3- or 2-position of the phenyl ring appeared to be well tolerated by GluK3, good examples of which are compounds 5, 19 and 21, which are equipotent or slightly more potent compared to the reference compound 1.
Analysis of collected data, with particular emphasis on the high GluK3 potency of 2- and 4-amino derivatives 21 and 23 (Ki = 1.0 and 1.4 µM, respectively), has led to the design of the third subseries of compounds that contained a heteroaromatic moiety in the ethynyl substituent (25–29). This modification appeared to be more preferable for binding to GluK3 receptors and yielded the most interesting compounds within the entire series, 27–29, showing 10-23-fold lower Ki at GluK3 compared to 1. Among analogues 27–29, compound 28 presented the most promising selectivity profile of all synthesized compounds, with submicromolar GluK3 affinity (Ki = 0.25 μM) and a preference of at least 400 times for homomeric receptors GluK3 over all other receptors tested.
The introduction of a heterocyclic substituent in the case of 27 and 29 resulted in submicromolar affinities at GluK3 receptors but with lower iGluR selectivity compared to 28. Compound 29, the most potent at GluK3 receptors, also showed a high binding affinity to the GluK2 and GluK1 KAR subtypes (Ki = 0.091 and 0.15 µM, respectively) as well as to the GluA2 AMPAR subtype (Ki = 0.23 µM).
3.2. Functional Pharmacology
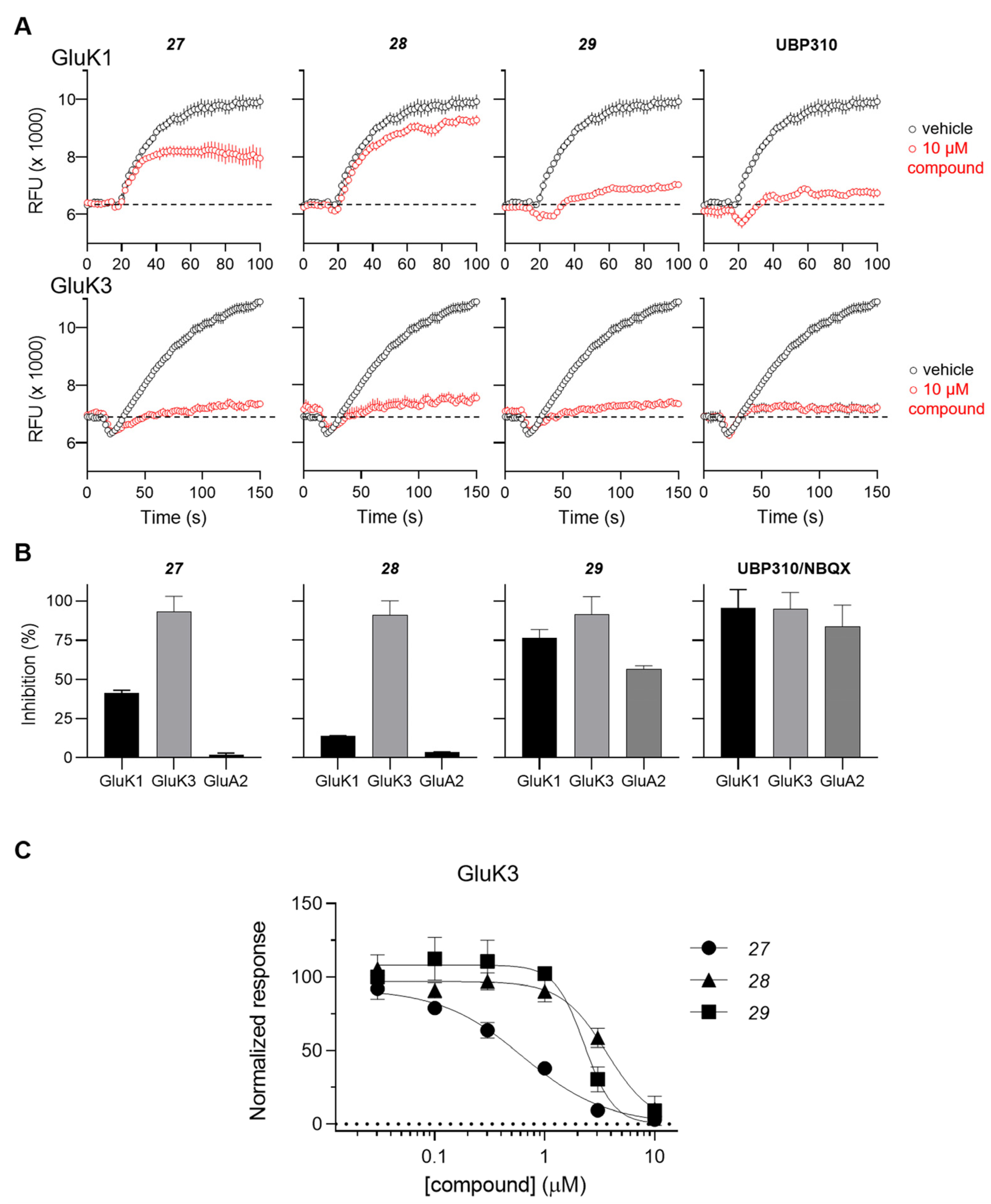
The antagonist properties of compounds 27–29 were confirmed at the GluK3 homomeric receptor subtype in an intracellular Ca2+ imaging assay. As shown in Figure 2, 27, 28 and 29 dose-dependently antagonized agonist-evoked responses at GluK3 with calculated IC50 values of 0.6, 3.6, and 2.2 μM, respectively.
Figure 2. Functional testing of 27, 28, and 29 at GluK1, GluK3 and GluA2 receptors in a Ca2+ fluorescence assay. (A) Characterization of antagonistic activity at GluK1 and GluK3 receptors using a 96-well based [Ca2+] imaging assay. Representative traces from recordings of Ca2+ dye fluorescence from HEK293 cells expressing GluK1 (upper traces) and GluK3 (lower traces) in absence (black) and presence (red) of the test compound. At t = 16 s, agonist-induced responses are evoked by application of 0.1 mM KA. The stippled lines indicate baseline fluorescence. Traces represent mean fluorescence from four identical wells. Error bars are the SEM and are shown when larger than symbol size. (B) Summary of the inhibition of agonist-evoked responses produced by 10 µM concentrations of the test compounds at GluK1, GluK3, and GluA2 receptors. The inhibition by the reference antagonists UBP310 (GluK1 and GluK3) and NBQX (GluA2) are shown to the right. Data points represent the mean from at eight individual wells. Error bars are the SEM and are shown when larger than symbol size. (C) Concentration inhibition curves for 27, 28, 29 at GluK3. Data points represent the mean from at least four individual wells. Error bars are the SEM and are shown when larger than symbol size. Maximum compound test concentration was limited to 10 μM due to the solubility issues of all compounds in the FLUO buffer at room temperature.
The inhibitory activity of 10 µM concentration of compounds 27, 28 and 29 was also tested at the GluK1 receptor and the AMPA-type GluA2 receptors. Compound 28 showed the highest level of selectivity by displaying less than 5% inhibition at GluK1 and GluA2 compared to full inhibition at GluK3 (Figure 2A,B).
4. Molecular Modeling
To investigate the binding mode of the new compounds and to elucidate the differences in ligand–protein interactions underlying the observed GluK3/GluK1 selectivity, molecular modeling studies have been performed. In the docking and molecular dynamics simulations, researchers used the available X-ray structure of the GluK1 ligand-binding domain (LBD) in complex with one of the quinoxaline-2,3-dione-based antagonists, PDB code 6SBT.
Flexible docking of all compounds to both the 6SBT structure and the GluK3-LBD homology model has been performed in the Schrodinger Suite environment
[33]. The predominant number of compounds was successfully docked to GluK1- and GluK3-LBD, with the top-ranking docking poses adopting a similar position inside both binding sites. The same characteristic pattern of interactions between the quinoxaline-2,3-dione scaffold and the D1 lobe of the receptor was observed for all ligands anchored, as in the case of other analogues in this chemical group, which were bound to the KAR or AMPAR subtypes
[32][34][35]. A crucial role in these interactions is played by Arg523/525, Pro516/518, and Thr518/520 (
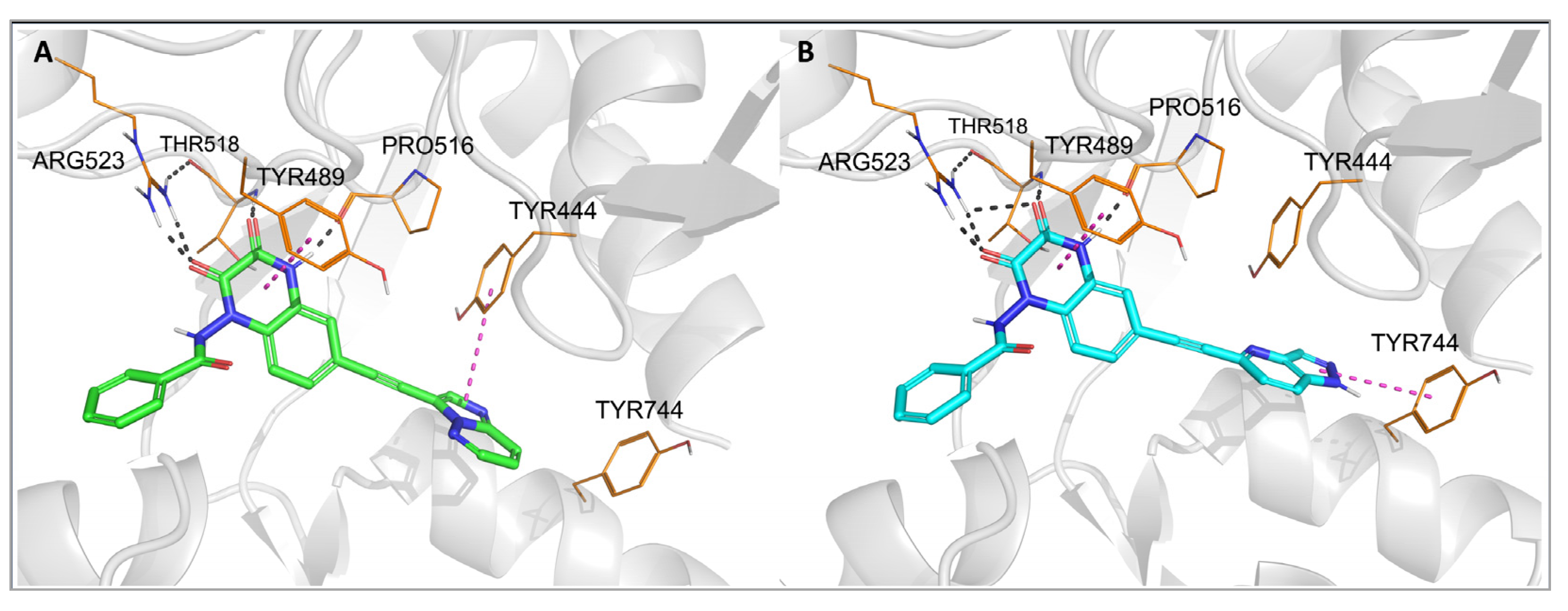
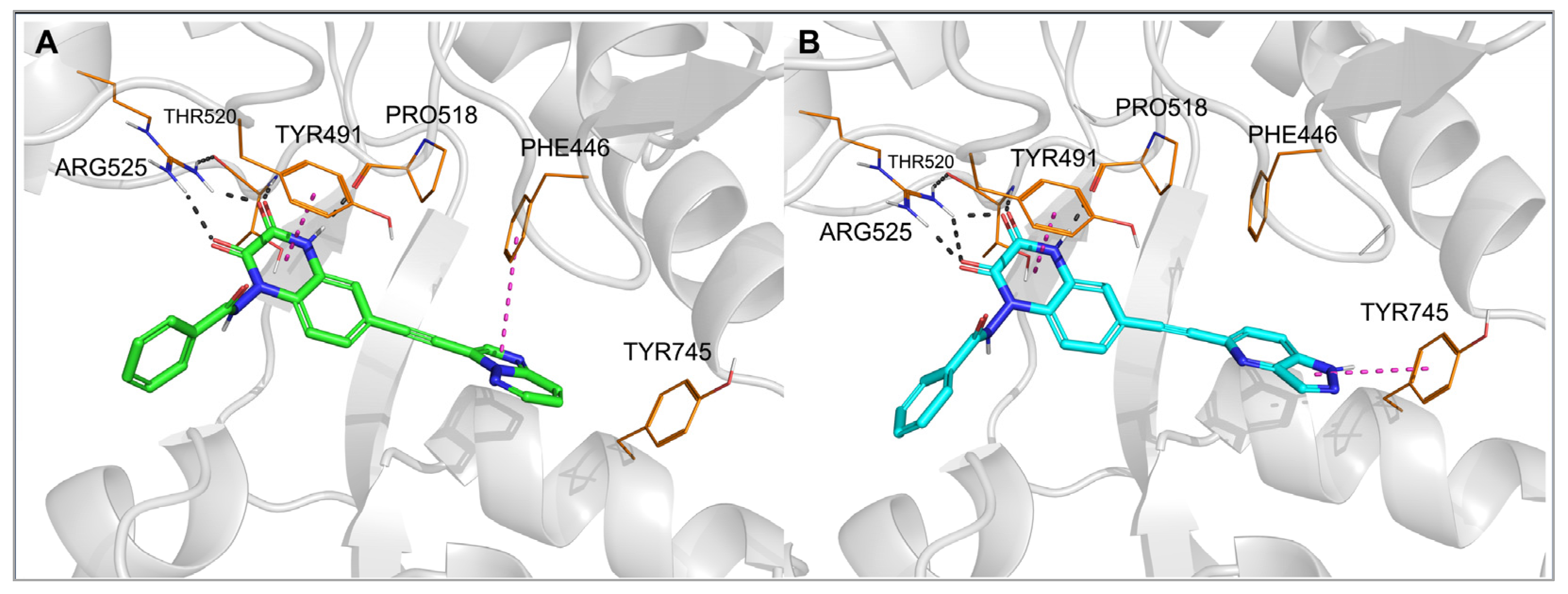
Figure 3 and
Figure 4, numbering of amino acids for GluK1 and GluK3 sequences, respectively). These amino acids are conserved among all KAR and AMPAR subunits. Additionally, the quinoxalinedione system was involved in π–π stacking with the aromatic ring of Tyr489/491. On the other hand, the benzamide moiety of the molecules was located at the border of GluK1 or GluK3 binding pockets, between the D1 and D2 lobes, and, for most of the ligands, did not form any direct interactions with the protein.
Figure 3. Top-ranked docking poses of 28 (A) and 29 (B) in the GluK1-LBD (PDB code: 6SBT). Hydrogen bonds are marked with gray dashes, π–π interactions are marked with pink dashes.
Figure 4. Top-ranked docking poses of 28 (A) and 29 (B) in the GluK3-LBD homology model. Hydrogen bonds are marked with gray dashes; π–π interactions are marked with pink dashes.
The observed GluK3-preference of the studied compounds was obviously determined by the substituent at position 6 of the quinoxaline-2,3-dione scaffold, as previously suggested
[32]. In the docking results, the arylethynyl substituent of the ligands was located near Tyr444/Ph446 and Tyr744/745, showing in most of the docking poses T-shaped π-stacking interactions with at least one of these residues (
Figure 3 and
Figure 4). However, in the case of
28, no direct interaction was found that could clearly discriminate between both receptors and explain the preferential binding of
28 to the GluK3 subunit.
For other X-ray structures of GluK1 antagonist complexes, 6FZ4 and 3S2V, it can be observed that the hydroxyl group of Tyr444 is involved in the water-mediated hydrogen bond network formed with the OH groups of Thr740 and Ser741
[32]. In the 4QF9 crystal structure, in turn, the alpha-amino acid moiety at position 6 of the quinoxalinedione replaces the water molecules, interacting with all the residues mentioned
[36]. In the case of most of the new derivatives described herein, an arylethynyl substituent at position 6 is probably not capable of contributing to the existing H-bond network system in GluK1, but it can disrupt it, which could be an explanation for the lack of affinity at this receptor. However, it should be noted that the GluK3-binding pocket in the vicinity of the arylethynyl tail of the docked ligands is more hydrophobic due to the replacement of Tyr444 and Ser741 in GluK1 for Phe446 and Thr742 in GluK3.
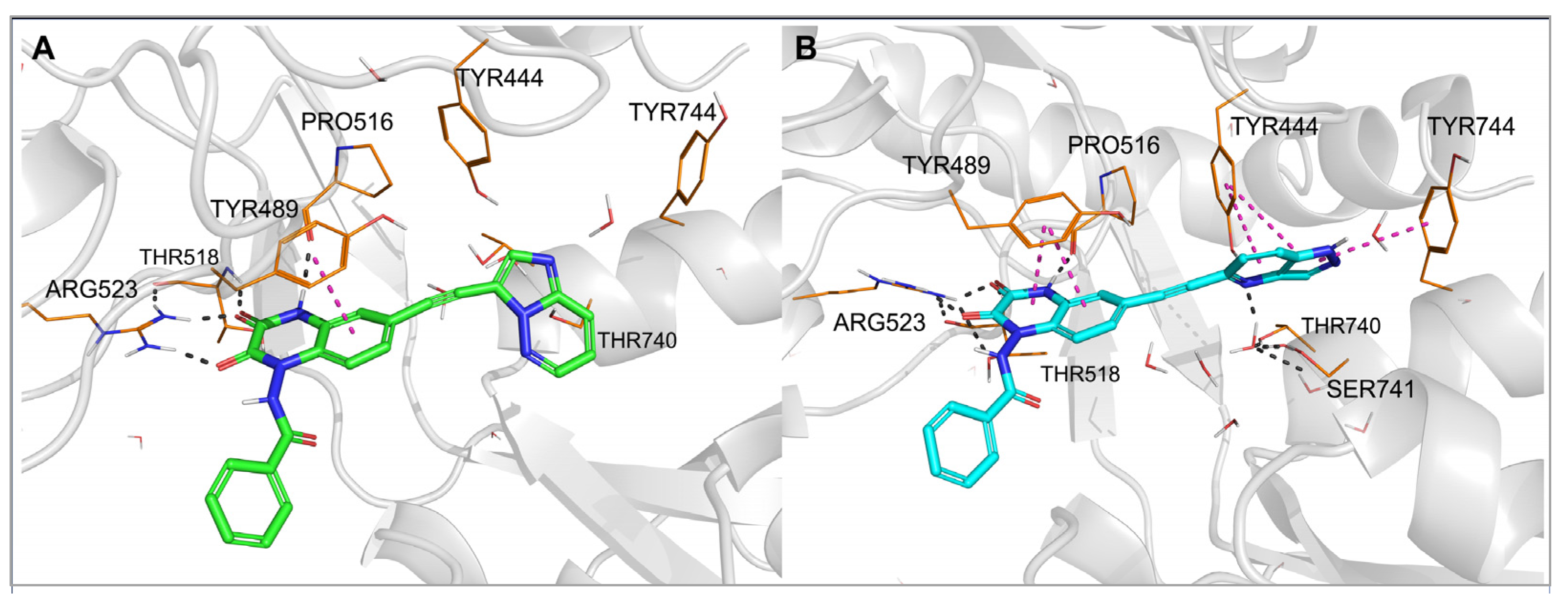
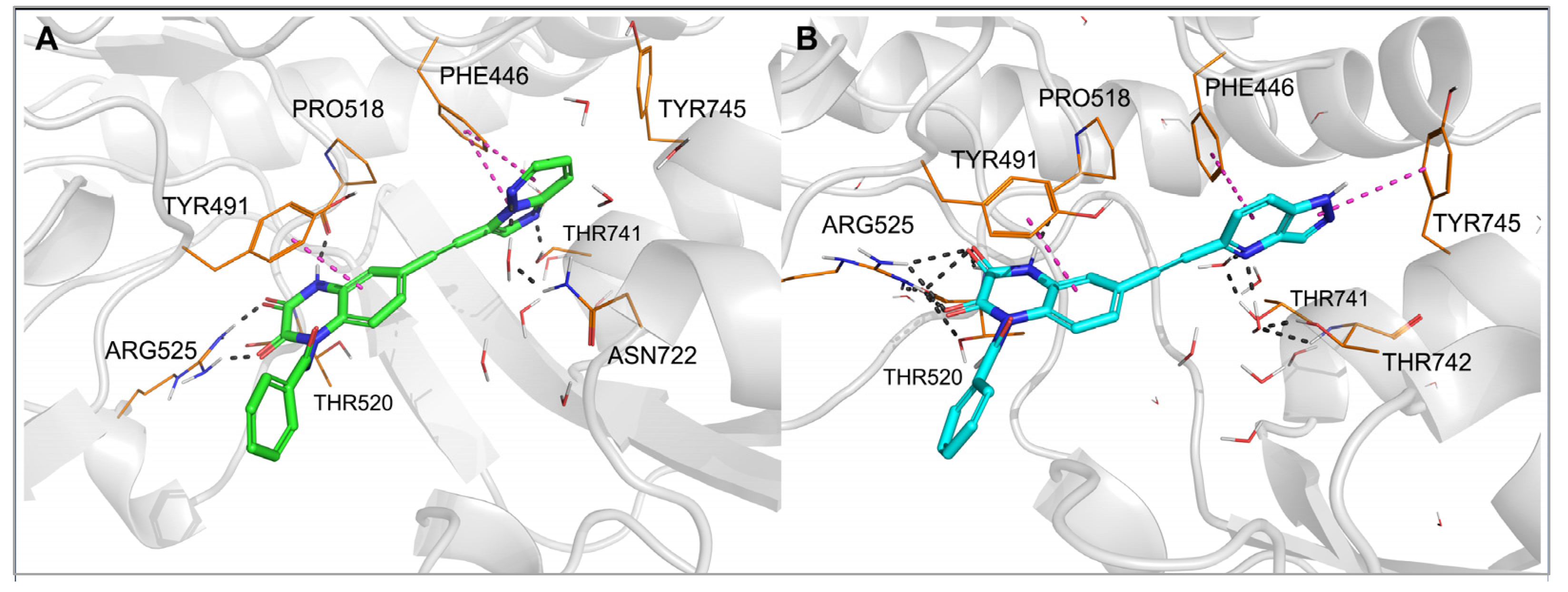
To examine in detail the molecular determinants underlying the observed affinity profile of 28 and 29, for individual complexes of those compounds with GluK1- and GluK3-LBD, previously obtained in flexible docking, molecular dynamics (MD) simulations were conducted (Figure 5 and Figure 6).
Figure 5. Complexes of the GluK1-LBD with compound 28 (A) and 29 (B), after 20 ns molecular dynamics simulation. Hydrogen bonds are marked with dark dashes, aromatic interactions are marked with pink dashes.
Figure 6. Complexes of the GluK3-LBD with compound 28 (A) and 29 (B) after 20 ns molecular dynamics simulation. Hydrogen bonds are marked with dark dashes, aromatic interactions are marked with pink dashes.
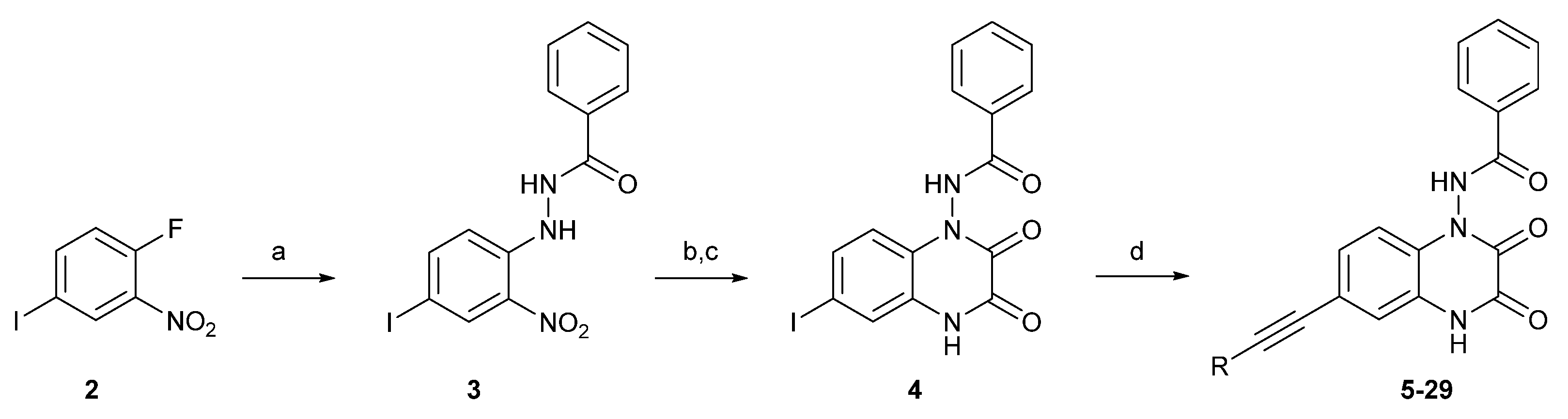
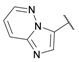
In each of the two MD simulations for 29-GluK1-LBD, rotation of the bicyclic aromatic moiety attached to the triple bond was observed compared to the initial docking pose. This allowed the nitrogen atom at position 4 of the 1H-pyrazolo [4,3-b] pyridin-5-yl fragment to create a hydrogen bond with the hydroxyl group of Tyr444 (Figure 5B), and at the same time, it did not affect the water-mediated H-bond network arranged between Tyr444, Ser741 and Thr740. Additionally, a stable CH–π stacking between the pyridine ring and Tyr444 as well as NH–π interaction between the pyrazole ring and Tyr744 were formed. Maintenance of a hydrogen bond network, in which the N4-nitrogen atom of the pyrazolopyridinyl moiety can replace one of the water molecules, with the simultaneous formation of favorable aromatic interactions, is likely to explain the high affinity of this compound for GluK1 receptors.
Another situation could be seen for 28, which is inactive at GluK1 but shows high potency and selectivity on the GluK3 receptor subunit. At the beginning of the MD simulation for the 28-GluK1-LBD complex, the imidazo [1,2-b]pyridazin-3-yl fragment moved away from the side chain of Tyr444 and Thr740 compared to the initial docking position, allowing access of water molecules to the hydroxyl groups of these residues and reconstitution of the H-bond network (Figure 5A). Displacement of the aromatic fragment from its initial location deprived it of the beneficial CH–π stacking with Tyr444. No other direct interactions were detected between this moiety and Tyr444, Thr740 or Tyr744, either. These results suggest that the substituent at position 6 of 28 does not fit the described GluK1 binding pocket and most likely explains its lack of activity at GluK1.
In the case of the 28-GluK3-LBD and 29-GluK3-LBD complexes, the results of the MD simulations were consistent for both compounds. The aromatic part of the arylethynyl substituent formed a stable CH–π stacking with Phe446 (Figure 6). The more extended pyrazolopyridinyl fragment of 29 allowed the NH–π stacking with Tyr745, while 28 formed hydrophobic interactions with this residue. The N4-nitrogen atom of the heterocyclic moiety in 29 created water-mediated H-bonds with Thr741 and Thr742 (Figure 6B). The N2-nitrogen atom of the imidazopyridazinyl fragment in 28 formed a direct hydrogen bond with the -OH group of Thr741. Furthermore, the position of the heterocyclic moiety in 28 allows for the creation of a water-mediated hydrogen bond between the N4-nitrogen atom and non-conserved Asn722 (Figure 6A). This amino acid is replaced in GluK1 by Ser721, with a shorter side chain, probably preventing the formation of an analogous interaction in GluK1. Therefore, the water-mediated hydrogen bond to Asn722 may be one of the determinants of the observed GluK3-selectivity of 28 in addition to the poor fit to GluK1 described above.
 +1 credit
+1 credit