Kainate receptors belong to the family of glutamate receptors ion channels, which are responsible for the majority of rapid excitatory synaptic transmission in the central nervous system. The therapeutic potential of kainate receptors is still poorly understood, which is also due to the lack of potent and subunit-selective pharmacological tools. In search of selective ligands for the GluK3 kainate receptor subtype, a series of quinoxaline-2,3-dione analogues was synthesized and pharmacologically characterized at selected recombinant ionotropic glutamate receptors. Among them, compound 28 (Table 1) was found to be a competitive GluK3 antagonist with submicromolar affinity and unprecedented high binding selectivity, showing a 400-fold preference for GluK3 over other homomeric receptors GluK1, GluK2, GluK5 and GluA2.
- glutamate receptors
- kainate receptors
- subunit selectivity
1. Introduction
2. Chemistry
The synthesis of the target quinoxaline-2,3-diones (5–29) was achieved through the synthetic route presented in Scheme 1, following a modified procedure described by Pallesen for compound 1 [33][32]. Commercially available 1-fluoro-4-iodo-2-nitrobenzene 2 was transformed into 3 through an SNAr reaction using benzohydrazide. The next step proceeded as a one-pot procedure through acylation, reduction of the nitro group, and spontaneous ring closure. The Sonogashira cross-coupling reaction allowed the introduction of a triple C-C bond moiety in place of iodine and the obtaining of the target structures (5–29).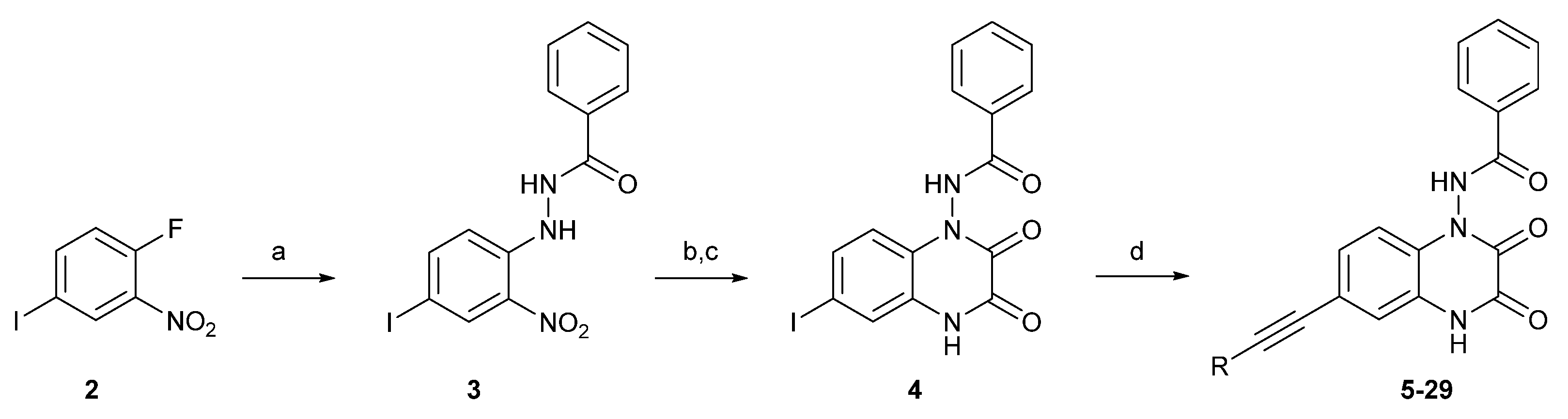
3. Pharmacological Characterization
3.1. Binding Pharmacology
The final compounds were characterized in binding studies using homomeric recombinant rat iGluRs: GluK1-GluK3, GluK5, and GluA2, expressed in Sf9 insect cell membranes. The in vitro binding data are presented in Table 1.| cmpd | R | Ki (µM) | ||||
|---|---|---|---|---|---|---|
| GluK1 | GluK2 | GluK3 | GluK5 | GluA2 | ||
| DNQX 1 | 0.65 ± 0.03 | 2.1 ± 0.3 | 0.36 ± 0.03 | 7.1 ± 0.9 | 0.25 ± 0.01 | |
| NBQX | 2.6 ± 0.1 | 5.4 ± 1.2 | 3.4 ± 0.6 | 152 ± 23 | 0.077 ± 0.010 | |
| 1 2 | Ph | >100 | ≈100 | 2.9 ± 0.3 | >100 | 24 ± 6 |
| 5 | 3-Me-Ph | ≈100 | ≈100 | 1.4 ± 0.4 | >100 | >100 |
| 6 | 4-Me-Ph | ≥100 | nd | 6.1 ± 0.8 | nd | nd |
| 7 | 4-Et-Ph | >100 | >100 | 20 ± 1 | >100 | >100 |
| 8 | 4-nPr-Ph | >100 | >100 | 50 ± 12 | >100 | >100 |
| 9 | 4-iPr-Ph | >100 | >100 | 38 ± 10 | >100 | >100 |
| 10 | 4-Cl-Ph | >100 | >100 | 3.5 ± 0.7 | >100 | >100 |
| 11 | 4-CF3-Ph | ≈100 | >100 | >100 | >100 | >100 |
| 12 | 4-MeO-Ph | >100 | >100 | 8.3 ± 0.4 | >100 | >100 |
| 13 | 4-PhO-Ph | ≥100 | nd | 7.9 ± 0.7 | nd | nd |
| 14 | 3-COOH-Ph | >100 | >100 | 28 ± 2 | >100 | >100 |
| 15 | 4-COOH-Ph | >100 | >100 | 31 ± 4 | >100 | >100 |
| 16 | 4-NH2SO2-Ph | >100 | >100 | ≈100 | >100 | >100 |
| 17 | 4-FSO2-Ph | 1.1 ± 0.4 | >100 | 8.1 ± 0.4 | >100 | ≈100 |
| 18 | 2-OH-Ph | >100 | nd | 14 ± 2 | nd | nd |
| 19 | 3-OH-Ph | >100 | >100 | 3.2 ± 0.6 | >100 | ≈100 |
| 20 | 4-OHCH2-Ph | ≈100 | >100 | 5.8 ± 1.5 | >100 | >100 |
| 21 | 2-NH2-Ph | 14 ± 2 | nd | 1.0 ± 0.1 | nd | nd |
| 22 | 3-NH2-Ph | >100 | nd | ≈100 | nd | nd |
| 23 | 4-NH2-Ph | >100 | >100 | 1.4 ± 0.2 | >100 | >100 |
| 24 | 4-N(CH3)2-Ph | 39 ± 4 | nd | 24 ± 4 | nd | nd |
| 25 | pyridin-4-yl | >100 | >100 | 25 ± 6 | >100 | >100 |
| 26 | 6-hydroxypyridin-3-yl | >100 | >100 | 11 ± 1 | >100 | >100 |
| 27 | pyrimidin-5-yl | 11 ± 2 | 12 ± 2 | 0.28 ± 0.02 | >100 | >100 |
| 28 | 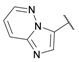 |
≈100 | >100 | 0.25 ± 0.01 | >100 | >100 |
| 29 | 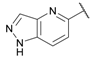 |
0.15 ± 0.05 | 0.091 ± 0.015 | 0.13 ± 0.03 | 3.8 ± 0.1 | 0.23 ± 0.02 |
3.2. Functional Pharmacology
The antagonist properties of compounds 27–29 were confirmed at the GluK3 homomeric receptor subtype in an intracellular Ca2+ imaging assay. As shown in Figure 2, 27, 28 and 29 dose-dependently antagonized agonist-evoked responses at GluK3 with calculated IC50 values of 0.6, 3.6, and 2.2 μM, respectively.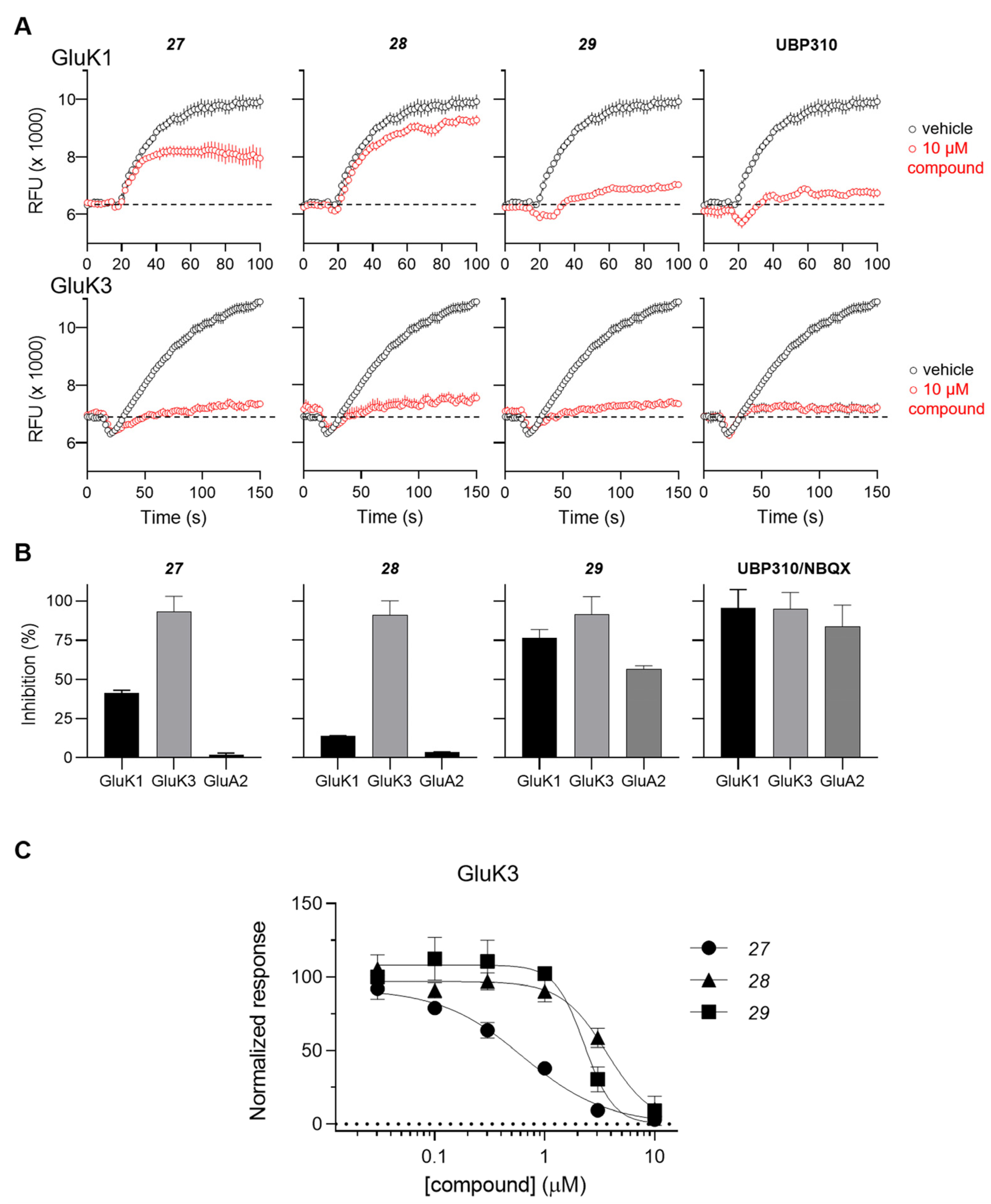
4. Molecular Modeling
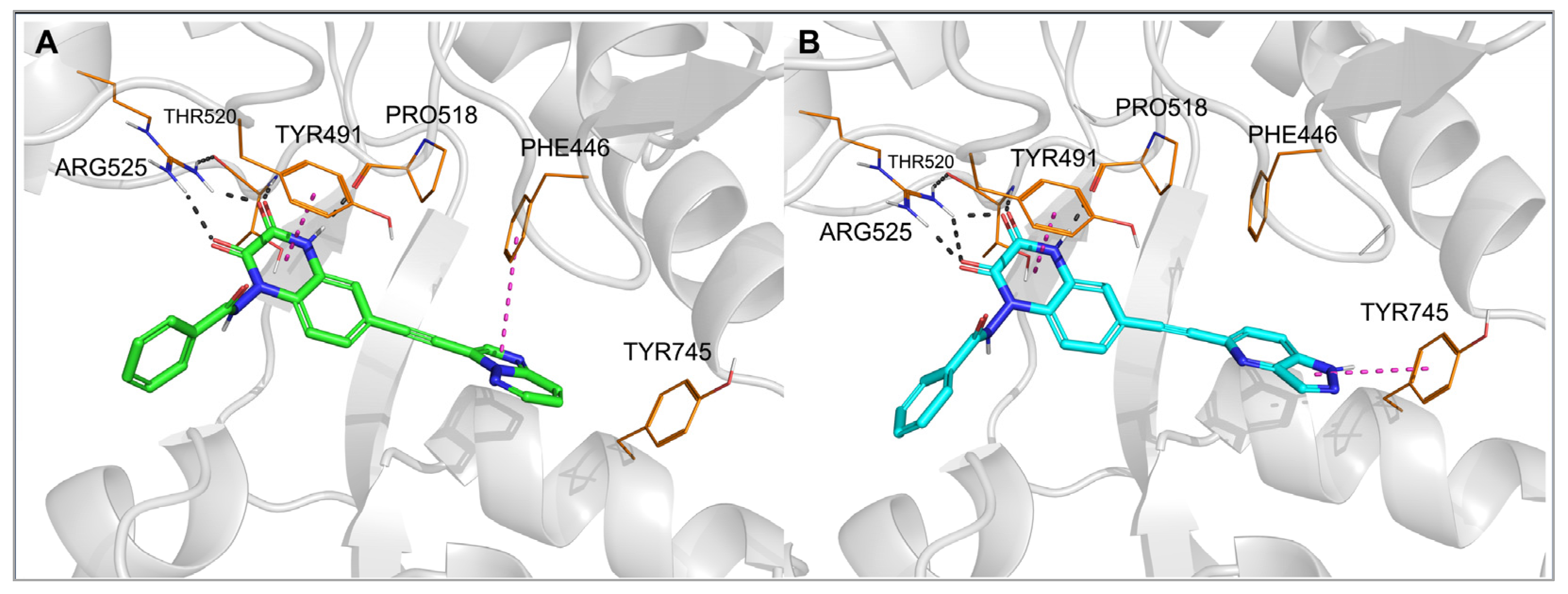
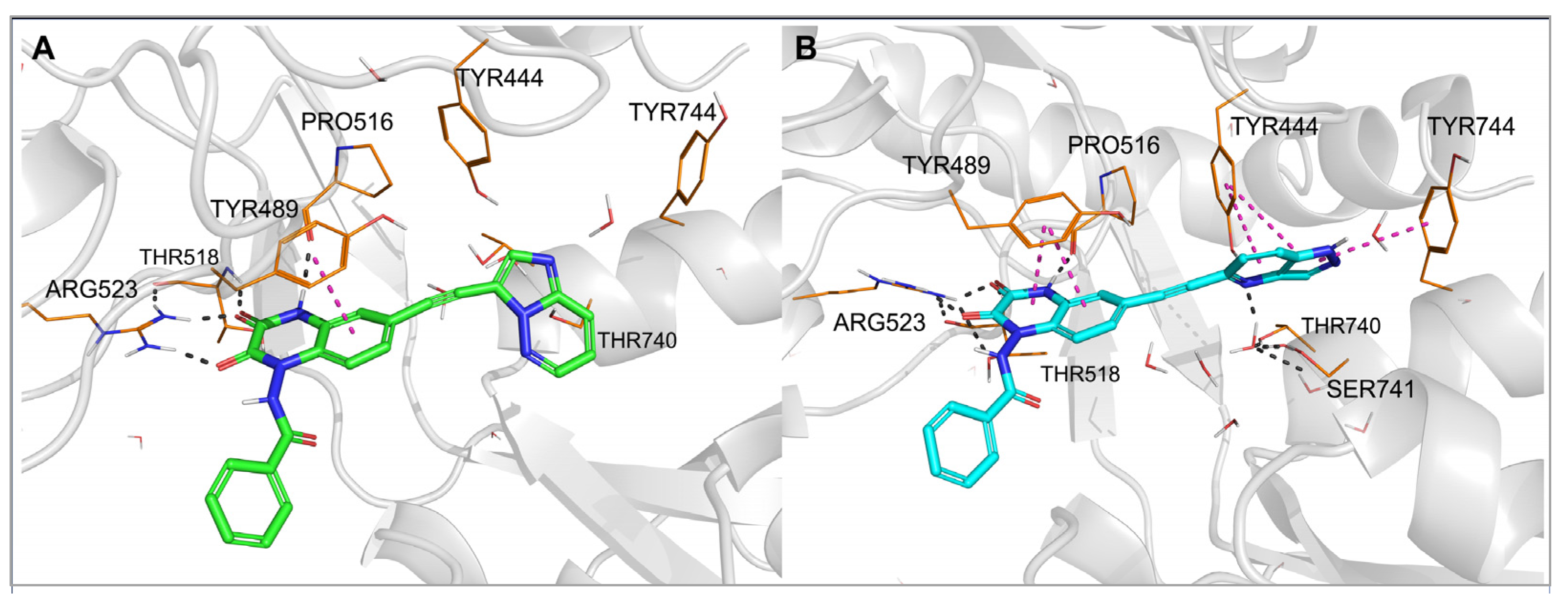
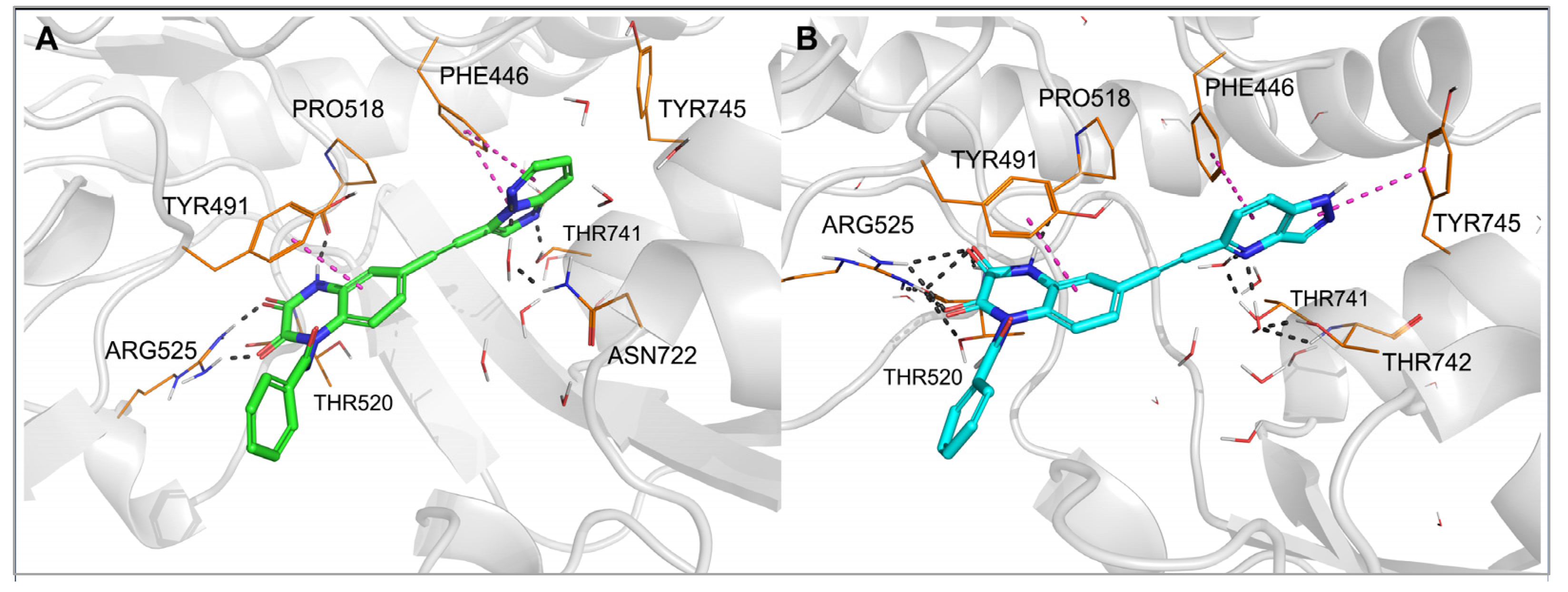
To investigate the binding mode of the new compounds and to elucidate the differences in ligand–protein interactions underlying the observed GluK3/GluK1 selectivity, molecular modeling studies have been performed. In the docking and molecular dynamics simulations, researchers used the available X-ray structure of the GluK1 ligand-binding domain (LBD) in complex with one of the quinoxaline-2,3-dione-based antagonists, PDB code 6SBT. Flexible docking of all compounds to both the 6SBT structure and the GluK3-LBD homology model has been performed in the Schrodinger Suite environment [35][33]. The predominant number of compounds was successfully docked to GluK1- and GluK3-LBD, with the top-ranking docking poses adopting a similar position inside both binding sites. The same characteristic pattern of interactions between the quinoxaline-2,3-dione scaffold and the D1 lobe of the receptor was observed for all ligands anchored, as in the case of other analogues in this chemical group, which were bound to the KAR or AMPAR subtypes [32,33,36][32][34][35]. A crucial role in these interactions is played by Arg523/525, Pro516/518, and Thr518/520 (Figure 3 and Figure 4, numbering of amino acids for GluK1 and GluK3 sequences, respectively). These amino acids are conserved among all KAR and AMPAR subunits. Additionally, the quinoxalinedione system was involved in π–π stacking with the aromatic ring of Tyr489/491. On the other hand, the benzamide moiety of the molecules was located at the border of GluK1 or GluK3 binding pockets, between the D1 and D2 lobes, and, for most of the ligands, did not form any direct interactions with the protein.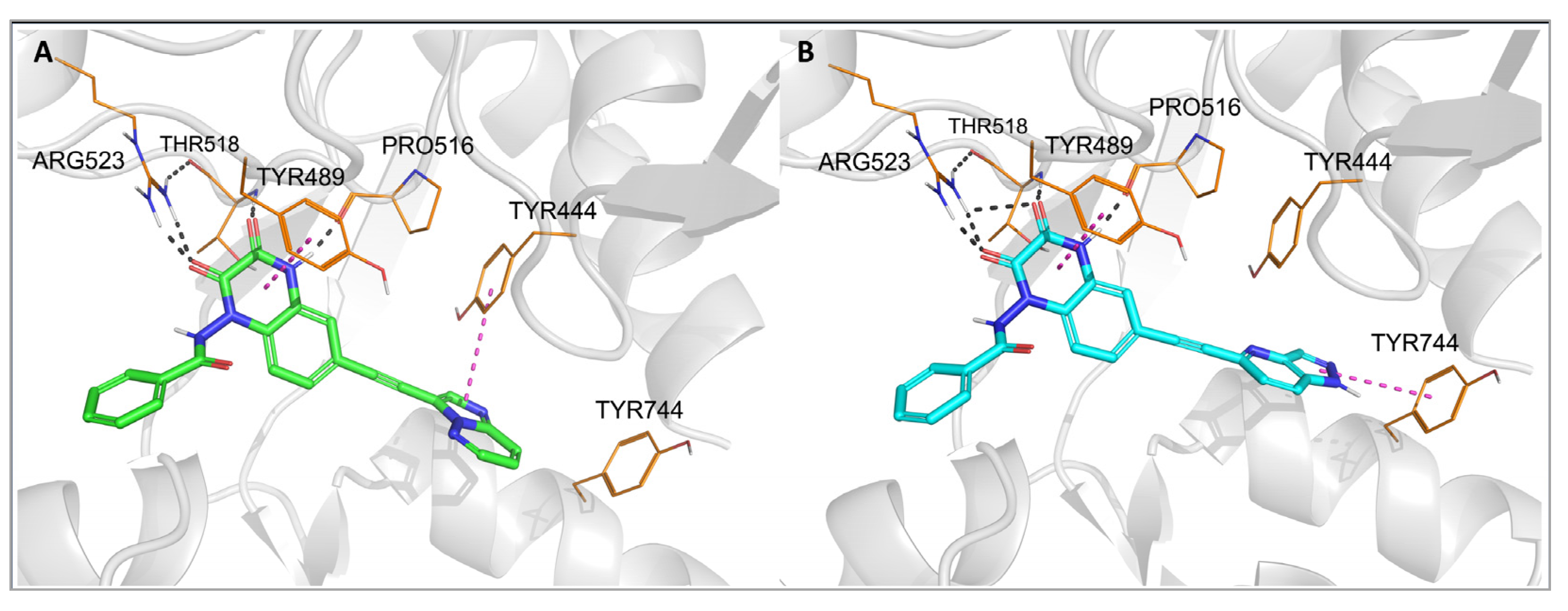
References
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol. Rev. 2021, 73, 298–487.
- Scholefield, C.L.; Atlason, P.T.; Jane, D.E.; Molnár, E. Assembly and trafficking of homomeric and heteromeric kainate receptors with impaired ligand binding sites. Neurochem. Res. 2019, 44, 585–599.
- Evans, A.J.; Gurung, S.; Henley, J.M.; Nakamura, Y.; Wilkinson, K.A. Exciting times: New advances towards nnderstanding the regulation and roles of kainate receptors. Neurochem. Res. 2019, 44, 572–584.
- Negrete-Díaz, J.V.; Falcón-Moya, R.; Rodríguez-Moreno, A. Kainate receptors: From synaptic activity to disease. FEBS J. 2021.
- Valbuena, S.; Lerma, J. Kainate receptors, homeostatic gatekeepers of synaptic plasticity. Neuroscience 2021, 456, 17–26.
- Pinheiro, P.S.; Perrais, D.; Coussen, F.; Barhanin, J.; Bettler, B.; Mann, J.R.; Malva, J.O.; Heinemann, S.F.; Mulle, C. GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc. Natl. Acad. Sci. USA 2007, 104, 12181–12186.
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311.
- Porter, R.H.; Eastwood, S.L.; Harrison, P.J. Distribution of kainate receptor subunit mRNAs in human hippocampus, neocortex and cerebellum, and bilateral reduction of hippocampal GluR6 and KA2 transcripts in schizophrenia. Brain Res. 1997, 751, 217–231.
- Takago, H.; Oshima-Takago, T. Pre- and postsynaptic ionotropic glutamate receptors in the auditory system of mammals. Hear. Res. 2018, 362, 1–13.
- Falcón-Moya, R.; Rodríguez-Moreno, A. Metabotropic actions of kainate receptors modulating glutamate release. Neuropharmacology 2021, 197, 108696.
- Negrete-Díaz, J.V.; Sihra, T.S.; Flores, G.; Rodríguez-Moreno, A. Non-canonical mechanisms of presynaptic kainate receptors controlling glutamate release. Front. Mol. Neurosci. 2018, 11, 128.
- Rodríguez-Moreno, A.; Sihra, T.S. Metabotropic actions of kainate receptors in the control of glutamate release in the hippocampus. Adv. Exp. Med. Biol. 2011, 717, 39–48.
- Sihra, T.S.; Rodríguez-Moreno, A. Presynaptic kainate receptor-mediated bidirectional modulatory actions: Mechanisms. Neurochem. Int. 2013, 62, 982–987.
- Perrais, D.; Pinheiro, P.S.; Jane, D.E.; Mulle, C. Antagonism of recombinant and native GluK3-containing kainate receptors. Neuropharmacology 2009, 56, 131–140.
- Falcón-Moya, R.; Sihra, T.S.; Rodríguez-Moreno, A. Kainate receptors: Role in epilepsy. Front. Mol. Neurosci. 2018, 11, 217.
- Fritsch, B.; Reis, J.; Gasior, M.; Kaminski, R.M.; Rogawski, M.A. Role of GluK1 kainate receptors in seizures, epileptic discharges, and epileptogenesis. J. Neurosci. 2014, 34, 5765–5775.
- Valbuena, S.; Lerma, J. Losing balance: Kainate receptors and psychiatric disorders comorbidities. Neuropharmacology 2021, 191, 108558.
- Zhuo, M. Cortical kainate receptors and behavioral anxiety. Mol. Brain 2017, 10, 16.
- Schiffer, H.H.; Heinemann, S.F. Association of the human kainate receptor GluR7 gene (GRIK3) with recurrent major depressive disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144b, 20–26.
- Ahmad, Y.; Bhatia, M.S.; Mediratta, P.K.; Sharma, K.K.; Negi, H.; Chosdol, K.; Sinha, S. Association between the ionotropic glutamate receptor kainate3 (GRIK3) Ser310Ala polymorphism and schizophrenia in the Indian population. World J. Biol. Psychiatry 2009, 10, 330–333.
- Kilic, G.; Ismail Kucukali, C.; Orhan, N.; Ozkok, E.; Zengin, A.; Aydin, M.; Kara, I. Are GRIK3 (T928G) gene variants in schizophrenia patients different from those in their first-degree relatives? Psychiatry Res. 2010, 175, 43–46.
- Samengo, I.; Curro, D.; Navarra, P.; Barrese, V.; Taglialatela, M.; Martire, M. Molecular and pharmacological evidence for a facilitatory functional role of pre-synaptic GLUK2/3 kainate receptors on GABA release in rat trigeminal caudal nucleus. Eur. J. Pain 2012, 16, 1148–1157.
- O’Neill, M.J.; Bogaert, L.; Hicks, C.A.; Bond, A.; Ward, M.A.; Ebinger, G.; Ornstein, P.L.; Michotte, Y.; Lodge, D. LY377770, a novel iGlu5 kainate receptor antagonist with neuroprotective effects in global and focal cerebral ischaemia. Neuropharmacology 2000, 39, 1575–1588.
- Weiss, B.; Alt, A.; Ogden, A.M.; Gates, M.; Dieckman, D.K.; Clemens-Smith, A.; Ho, K.H.; Jarvie, K.; Rizkalla, G.; Wright, R.A.; et al. Pharmacological characterization of the competitive GLUK5 receptor antagonist decahydroisoquinoline LY466195 in vitro and in vivo. J. Pharmacol. Exp. Ther. 2006, 318, 772–781.
- Lubisch, W.; Behl, B.; Henn, C.; Hofmann, H.P.; Reeb, J.; Regner, F.; Vierling, M. Pyrrolylquinoxalinediones carrying a piperazine residue represent highly potent and selective ligands to the homomeric kainate receptor GluR5. Bioorganic Med. Chem. Lett. 2002, 12, 2113–2116.
- Dolman, N.P.; More, J.C.; Alt, A.; Knauss, J.L.; Pentikainen, O.T.; Glasser, C.R.; Bleakman, D.; Mayer, M.L.; Collingridge, G.L.; Jane, D.E. Synthesis and pharmacological characterization of N3-substituted willardiine derivatives: Role of the substituent at the 5-position of the uracil ring in the development of highly potent and selective GLUK5 kainate receptor antagonists. J. Med. Chem. 2007, 50, 1558–1570.
- More, J.C.A.; Nistico, R.; Dolman, N.P.; Clarke, V.R.J.; Alt, A.J.; Ogden, A.M.; Buelens, F.P.; Troop, H.M.; Kelland, E.E.; Pilato, F.; et al. Characterisation of UBP296: A novel, potent and selective kainate receptor antagonist. Neuropharmacology 2004, 47, 46–64.
- Poulie, C.B.M.; Larsen, Y.; Leteneur, C.; Barthet, G.; Bjørn-Yoshimoto, W.E.; Malhaire, F.; Nielsen, B.; Pin, J.-P.; Mulle, C.; Pickering, D.S.; et al. (S)-2-Mercaptohistidine: A first selective orthosteric GluK3 antagonist. ACS Chem. Neurosci. 2022, 13, 1580–1587.
- Honoré, T.; Davies, S.N.; Drejer, J.; Fletcher, E.J.; Jacobsen, P.; Lodge, D.; Nielsen, F.E. Quinoxalinediones: Potent competitive non-NMDA glutamate receptor antagonists. Science 1988, 241, 701–703.
- Sheardown, M.J.; Nielsen, E.O.; Hansen, A.J.; Jacobsen, P.; Honore, T. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: A neuroprotectant for cerebral ischemia. Science 1990, 247, 571–574.
- Catarzi, D.; Colotta, V.; Varano, F. Competitive AMPA receptor antagonists. Med. Res. Rev. 2007, 27, 239–278.
- Pallesen, J.; Møllerud, S.; Frydenvang, K.; Pickering, D.S.; Bornholdt, J.; Nielsen, B.; Pasini, D.; Han, L.; Marconi, L.; Kastrup, J.S.; et al. N1-substituted quinoxaline-2,3-diones as kainate receptor antagonists: X-ray crystallography, structure−affinity relationships, and in vitro pharmacology. ACS Chem. Neurosci. 2019, 10, 1841–1853.
- Schrödinger Release 2021-4; LigPrep; Epik; Protein Preparation Wizard; Macromodel; Glide; Prime; MM-GBSA. Schrödinger, LLC.: New York, NY, USA, 2021.
- Møllerud, S.; Hansen, R.B.; Pallesen, J.; Temperini, P.; Pasini, D.; Bornholt, J.; Nielsen, B.; Mamedova, E.; Chalupnik, P.; Paternain, A.V.; et al. N-(7-(1 H-Imidazol-1-yl)-2,3-dioxo-6-(trifluoromethyl)-3,4-dihydroquinoxalin-1(2 H)-yl)benzamide, a new kainate receptor selective antagonist and analgesic: Synthesis, X-ray crystallography, structure-affinity relationships, and in vitro and in vivo pharmacology. ACS Chem. Neurosci. 2019, 10, 4685–4695.
- Pøhlsgaard, J.; Frydenvang, K.; Madsen, U.; Kastrup, J.S. Lessons from more than 80 structures of the GluA2 ligand-binding domain in complex with agonists, antagonists and allosteric modulators. Neuropharmacology 2011, 60, 135–150.
- Demmer, C.S.; Rombach, D.; Liu, N.; Nielsen, B.; Pickering, D.S.; Bunch, L. Revisiting the quinoxalinedione scaffold in the construction of new ligands for the ionotropic glutamate receptors. ACS Chem. Neurosci. 2017, 8, 2477–2495.