 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Krishnan Sriram | -- | 9342 | 2023-02-08 16:19:58 | | | |
| 2 | Camila Xu | Meta information modification | 9342 | 2023-02-09 03:14:59 | | |
Video Upload Options
An occupational injury describes any injury or illness to a worker as related to their specific work/occupational demands or requirement. It is often a life-altering event that is also considered a form of disability, which is covered under the provisions of the 1990 Americans with Disabilities Act. Occupational injuries and toxicant exposures, particularly to the brain, can lead to the development of neuroinflammation by activating distinct mechanistic signaling cascades that ultimately culminate in the disruption of neuronal function leading to neurological and neurodegenerative disorders. The entry of toxicants into the brain causes the subsequent activation of glial cells, a response known as ‘reactive gliosis.’ Gliosis, a “reactive” state of glia, is a pathological hallmark of all types of central nervous system (CNS) injuries. Advancements in the field of neuroscience, including techniques for high throughput gene expression analysis, morphometric analysis, cytokine and chemokine analysis, advances in neuroimaging and cellular microscopy, have helped identify the possible molecular and neural targets associated with occupational injury or toxicant exposure, bringing the neuroinflammation perspective into place for the implication of gliosis and subsequent progression of neural injury in conditions like TBI and neurodegenerative diseases like AD, PD, CJD, ALS, and MS.
1. Occupational Brain Injuries
2. Reactive Gliosis in Neural Injury and Neurodegeneration
| Toxicant/Injury/Disease | Brain Area(s) Affected | Reference(s) |
|---|---|---|
| Occupational Toxicants: | ||
| 1,1,1-trichloroethane | CTX, CER, HIP | [23] |
| Acrylamide | CER | [24] |
| Chlorpyrifos | STR | [25] |
| Cyclohexane | HIP | [26] |
| Dichloromethane | FCT, MCT | [27] |
| Fracking Sand Dust | OB, STR, CER | [28] |
| Manganese | GP, SN-R | [29] |
| Manganese | HIP | [30] |
| Oil dispersant (COREXIT EC9500A) | FCT, HIP | [31] |
| Silver nanoparticles | FCT, HIP | [32] |
| Toluene | HIP | [33] |
| Toluene | HIP, CER | [34] |
| Toluene | STR | [35] |
| Trimethyltin | HIP | [36] |
| Trimethyltin | HIP | [37] |
| Welding fumes | STR, MB | [38] |
| Welding fumes | STR, MB | [39] |
| Xylene | FCT, CER | [27] |
| Brain Injuries: | ||
| Cerebellar stab injury | CER | [40] |
| Cortical stab injury | CTX | [41] |
| Cortical stab injury | CTX | [42] |
| Facial nerve lesion | CTX | [43] |
| Forebrain stab lesion | HIP | [44] |
| Hippocampal stab wound | HIP | [45] |
| Severe focal brain ischemia | CTX | [46] |
| Transient global ischemia | HIP | [47] |
| Transient global ischemia | HIP | [48] |
| Transient MCAO | STR, HIP, CTX | [49] |
| Traumatic brain injury | MCT | [50] |
| Traumatic brain injury | CTX | [51] |
| Traumatic brain injury | HIP | [52] |
| Neurologic diseases/disorders: | ||
| Alzheimer’s disease | TCT | [53] |
| Alzheimer’s disease | HIP | [54] |
| Alzheimer’s disease | CN, THL, CER, BS | [55] |
| Alzheimer’s disease | HIP | [56] |
| Alzheimer’s disease | CTX, PUT, AMG | [57] |
| Alzheimer’s disease | HIP, FCT, PCT, TCT, CER | [58] |
| Alzheimer’s disease | ECT | [59] |
| Alzheimer’s-type dementia | TCT | [60] |
| Amyotrophic lateral sclerosis | CTX | [61] |
| Creutzfeldt-Jackob disease | CTX | [62] |
| Creutzfeldt-Jackob disease | OB, CTX, HIP, AMG | [63] |
| Multiple sclerosis | CTX | [64] |
| Multiple sclerosis | WM | [65] |
| Parkinson’s disease | SN, LC, DVN | [66] |
| Parkinson’s disease | OB | [67] |
| Schizophrenia with dementia | FCT, TCT | [68] |
| Toxicant/Injury/Disease | Brain Area(s) Affected | Reference(s) |
|---|---|---|
| Occupational Toxicants: | ||
| 2, 5-Hexanedione | SN | [69] |
| Acrylamide | SN | [70] |
| Aluminum | STR, HIP, CTX | [71] |
| Cypermethrin | STR, SN | [72] |
| Diacetyl | OB | [73] |
| Dichlorvos | STR, SN | [74] |
| Diesel Exhaust | OB, CTX, MB, SN | [75] |
| Diethyldithiocarbamate | ECT, HIP, HYP | [76] |
| Iron oxide nanoparticles | OB, HIP, STR | [77] |
| Manganese | SN | [78] |
| Manganese | GP | [79] |
| Paraquat + Maneb | LC | [80] |
| Rotenone | STR, SN | [81] |
| Rotenone | HIP, CTX | [82] |
| Silver nanoparticles | OB | [83] |
| Silver nanoparticles | OB | [84] |
| Trimethyltin | HIP | [85] |
| Trimethyltin | HIP | [37] |
| Welding fumes | STR, MB | [86] |
| Welding fumes | STR, MB | [87] |
| Brain Injuries: | ||
| Cortical stab injury | CTX | [42] |
| Mild focal brain ischemia | STR | [88] |
| Transient focal ischemia | SN | [89] |
| Transient global ischemia | STR, HIP | [90] |
| Transient MCAO | SN | [91] |
| Traumatic brain injury | CTX | [92] |
| Traumatic brain injury | CWM, THL | [93] |
| Traumatic brain injury | CTX | [51] |
| Traumatic brain injury | HIP | [52] |
| Neurologic diseases/disorders: | ||
| Alzheimer’s disease | FCT, TCT, OCT, CER, BS | [94] |
| Alzheimer’s disease | FCT | [95] |
| Alzheimer’s disease | ITG, MTG | [96] |
| Alzheimer’s disease | CER | [97] |
| Alzheimer’s disease | HIP | [98] |
| Alzheimer’s disease | FCT | [99] |
| Alzheimer-type dementia | ECT, CCT, TCT | [100] |
| Amyotrophic lateral sclerosis | CTX, HIP | [101] |
| Amyotrophic lateral sclerosis | FTCT | [102] |
| Amyotrophic lateral sclerosis | CC | [103] |
| Creutzfeldt-Jackob disease | FCT, TCT, STR, TH, CER | [104] |
| Multiple sclerosis | WM | [65] |
| Multiple system atrophy | STR | [105] |
| Parkinson’s disease | SN | [106] |
| Parkinson’s disease | SN | [107] |
| Viral Encephalitis | FCT, TCT, OCT, CER, BS | [94] |
| Wilson’s disease | FCT, TCT, OCT, CER, BS | [94] |
2.1. Gliosis in Brain Injury
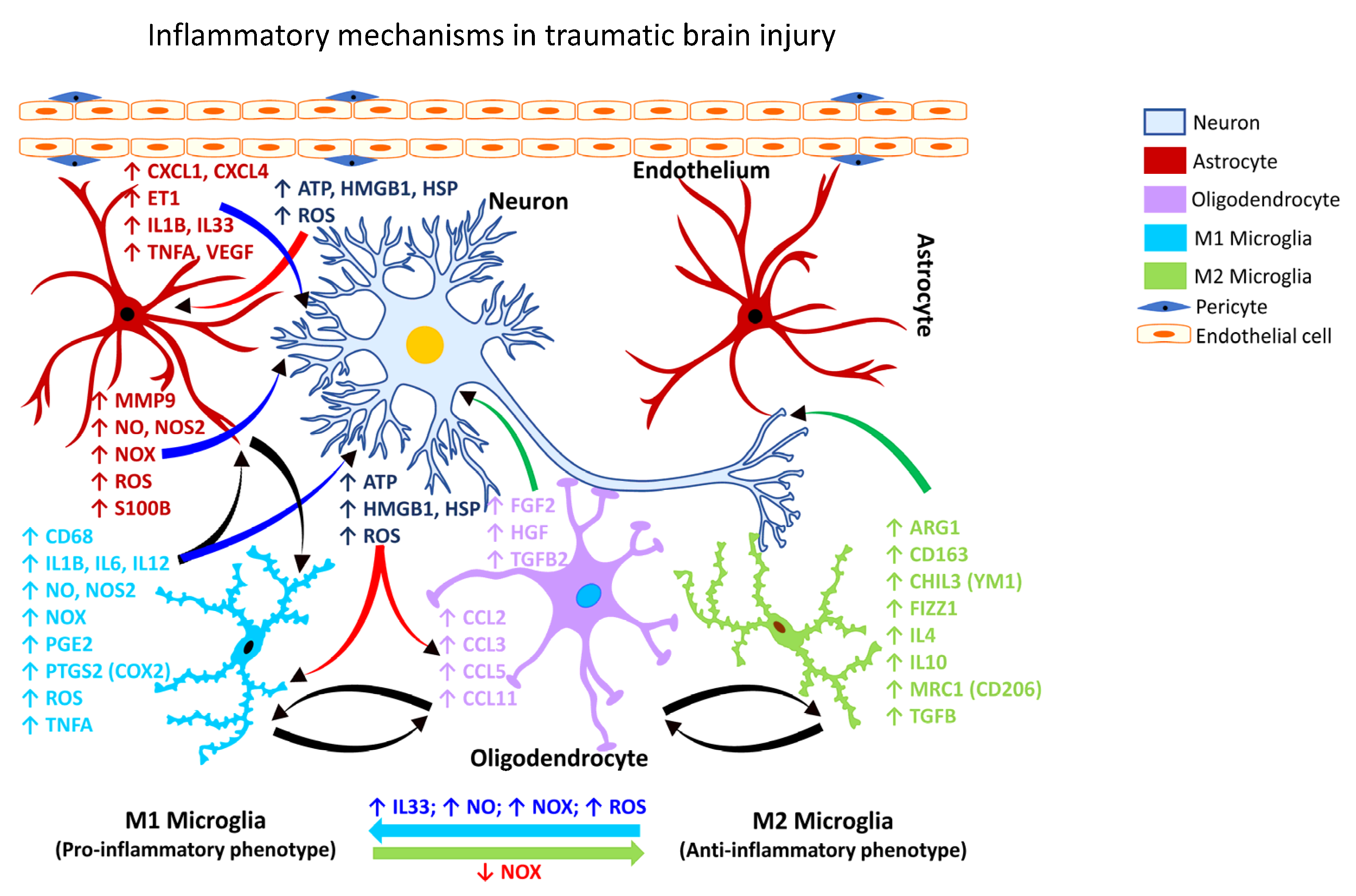
2.2. Gliosis Following Chemical-Induced Neural Injury
2.2.1. Gliosis Associated with Hydrocarbon Exposure
2.2.2. Gliosis Associated with Metal Exposure
2.2.3. Gliosis Associated with Nanoparticles/Nanomaterials
Carbon-Based Nanomaterials
Metal Oxide Nanoparticles
2.3. Gliosis in Neurological Disease States
2.3.1. Gliosis in AD
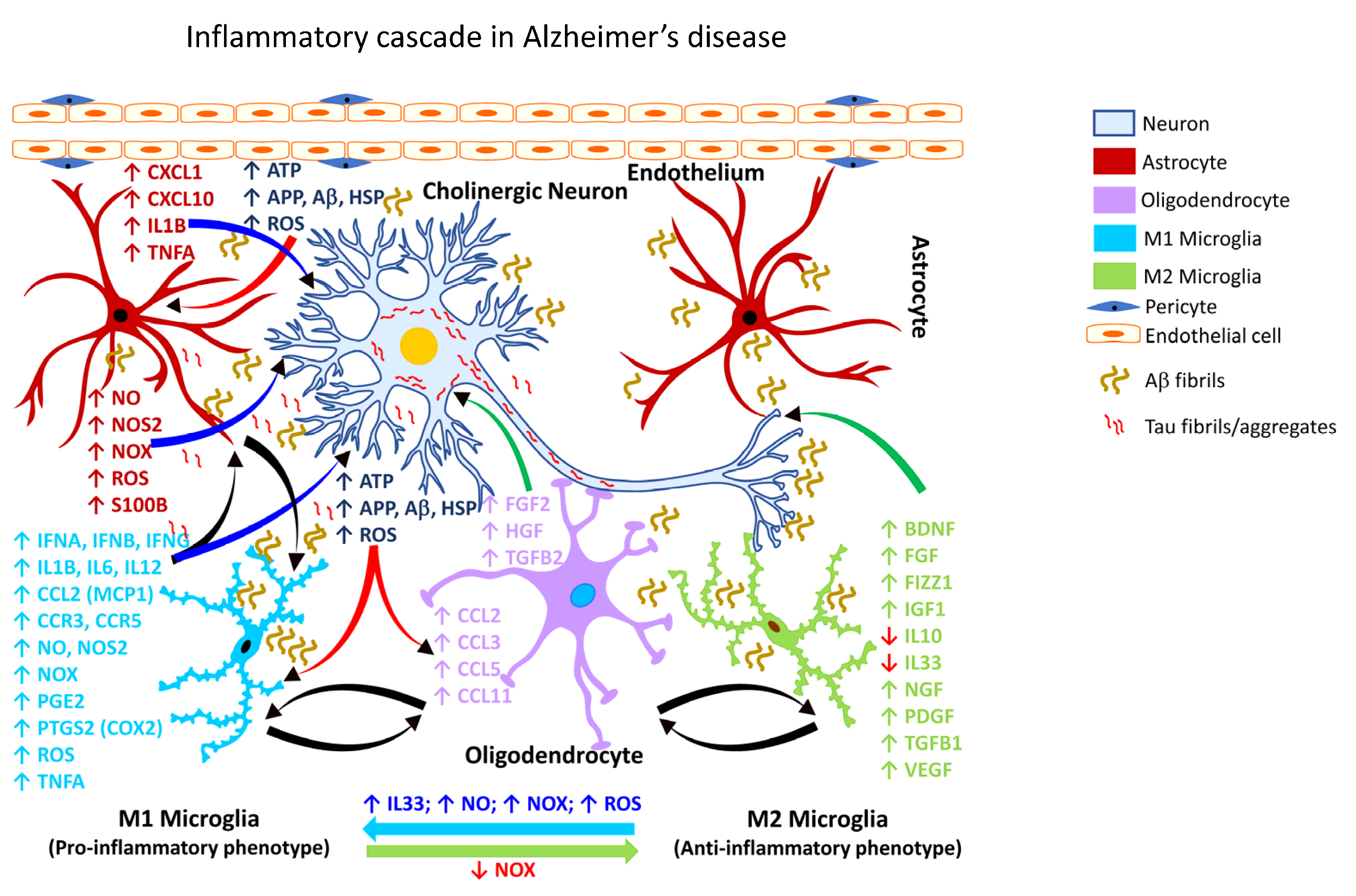
| Glial Cell Type | Inflammatory Mediator | Cellular (Neural) Effect | Reference(s) |
|---|---|---|---|
| M1 Microglia (reactive) Astrocyte (reactive) |
TNFA | ↑ Glutaminase ↑ Glutamic acid ↑ NO ↑ Peroxynitrite formation ↑ BACE 1 & 2 (Beta Secretases) ↑ APH1A (Gamma secretase) ↑ Aβ formation ↑ Aβ aggregation ↑ Aβ oligomers ↑ Aβ fibrils/plaques ↑ Aβ toxicity ↑ Tau ↑ P-Tau ↑ P-Tau ↑ P-Tau aggregates ↑ Tau fibrils/NFTs ↑ Synaptic excitotoxicity ↑ Synaptic dysfunction ↑ Neuronal Apoptosis ↓ Aβ clearance ↓ LTP |
[229][230][231][232][233][234][235] |
| M1 Microglia (reactive) Astrocyte (reactive) |
IL1B | ↑ APP expression ↑ Aβ formation ↑ Aβ aggregation ↑ Aβ oligomers ↑ Aβ fibrils/plaques ↑ Aβ toxicity ↑ Tau ↑ P-Tau ↑ P-Tau ↑ P-Tau aggregates ↑ Tau fibrils/NFTs ↑ NO ↑ Peroxynitrite formation ↑ S100B ↑ p38 MAPK ↑ PTGS2 (COX2) ↑ Prostaglandin E2 (PGE2) ↑ Synaptic plasticity ↑ Synaptic dysfunction ↑ Neuronal damage ↑ Neuronal death ↓ LTP |
[229][231][236][237][238][239][240][241][242][243][244] |
| M1 Microglia (reactive) Astrocyte (reactive) |
IL6 | ↑ APP expression ↑ Tau ↑ P-Tau ↑ P-Tau ↑ P-Tau aggregates ↑ Tau fibrils/NFTs ↑ p38 MAPK ↑ p35 activator ↑ CDK5 ↓ LTP |
[245][246][247][248][249] |
| M1 Microglia (reactive) Astrocyte (reactive) |
IL18 | ↑ Aβ formation ↑ Aβ aggregation ↑ Aβ oligomers ↑ Aβ fibrils/plaques ↑ Aβ toxicity ↑ Tau ↑ P-Tau ↑ P-Tau ↑ P-Tau aggregates ↑ Tau fibrils/NFTs ↑ Synaptic dysfunction ↑ Neuronal damage ↑ Neuronal death ↓ LTP |
[250][251][252] |
| M1 Microglia (reactive) Astrocyte (reactive) |
COX1 | ↑ Inflammation | [253] |
| M1 Microglia (reactive) Astrocyte (reactive) |
NO | ↑ Inflammation ↑ Protein oxidative damage ↑ Lipid oxidation ↑ Tau fibrils/NFTs ↑ Synaptic excitotoxicity ↑ Synaptic dysfunction ↑ Neuronal damage ↑ Neuronal death |
[254][255][256] |
2.3.2. Gliosis in PD
| Glial Cell Type | Inflammatory Mediator | Cellular (Neural) Effect | Reference(s) |
|---|---|---|---|
| M1 Microglia (reactive) Astrocyte (reactive) |
TNFA | ↑ SNCA fibrils ↑ SNCA aggregation ↑ Excitatory synaptic transmission ↑ AMPA receptors ↑ Synaptic dysfunction ↑ Neuronal apoptosis ↑ Neuronal degeneration ↑ mEPSC frequency ↑ mEPSC amplitude ↓ LTP ↓ Inhibitory synaptic transmission ↓ GABRA1 receptors ↓ mIPSC amplitude |
[275][276][277][278][279][280][281][282] |
| M1 Microglia (reactive) Astrocyte (reactive) |
IL1B | ↑ MAPK ↑ SRC Kinase ↑ SNCA fibrils ↑ Apoptosis ↑ Excitotoxicity ↑ Synaptic dysfunction ↑ Neuronal damage ↑ Neuronal death |
[283][284][285][286][287] |
| M1 Microglia (reactive) Astrocyte (reactive) |
TGFB | ↑ Deregulation of ligands ↑ Deregulation of receptors ↑ Neuronal damage ↑ Neuronal death |
[288][289][290][291] |
| M1 Microglia (reactive) Astrocyte (reactive) |
NO | ↑ Peroxynitrite formation ↑ S-Nitrosylation of proteins ↑ S-Nitrosylation of PARK proteins ↑ Excitotoxicity ↑ Impaired mitochondria ↑ Mitochondrial dysfunction ↑ Neuronal damage ↑ Neuronal death |
[292][293][294][295][296] |
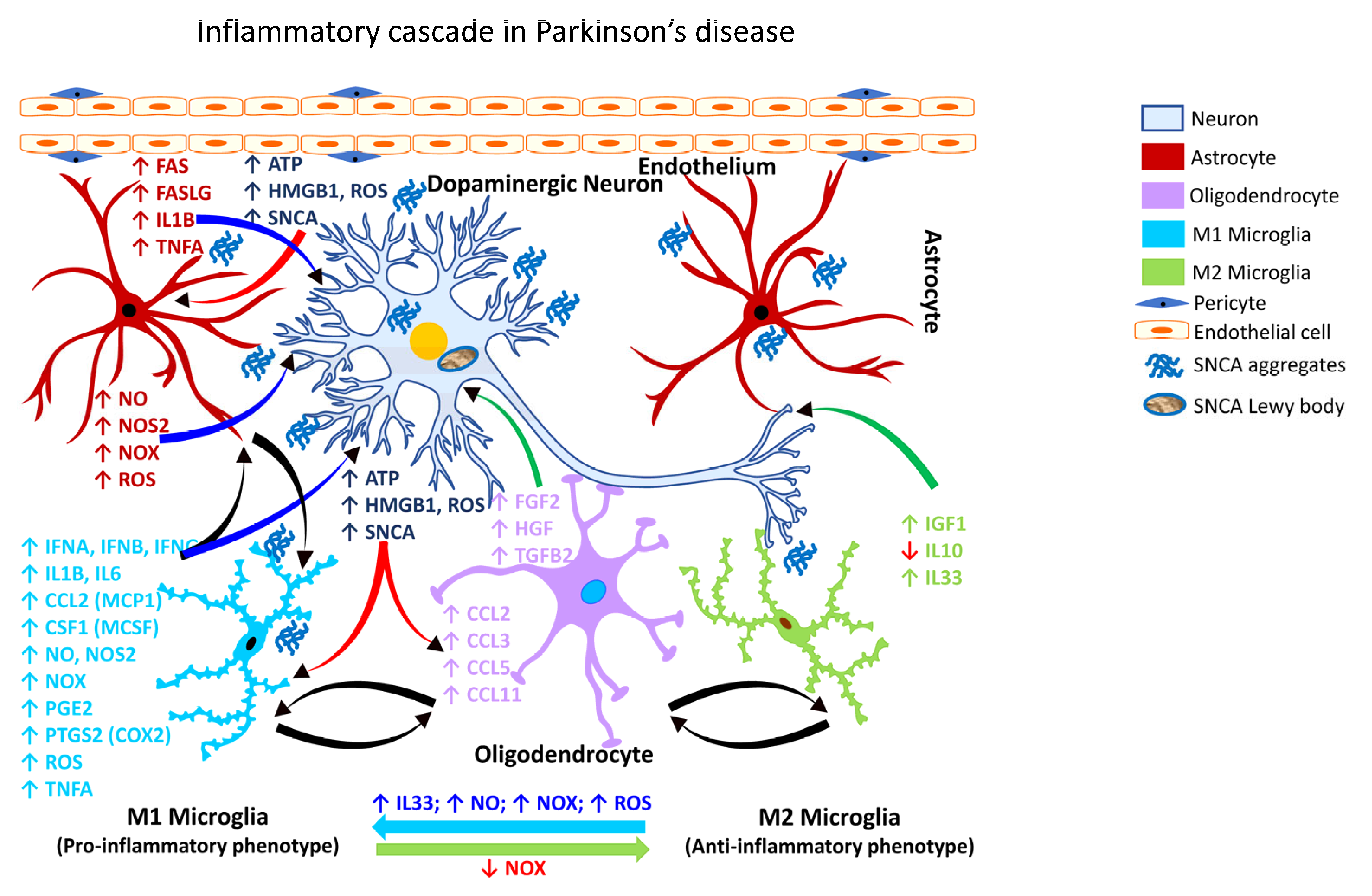
2.3.3. Gliosis in CJD
2.3.4. Gliosis in ALS
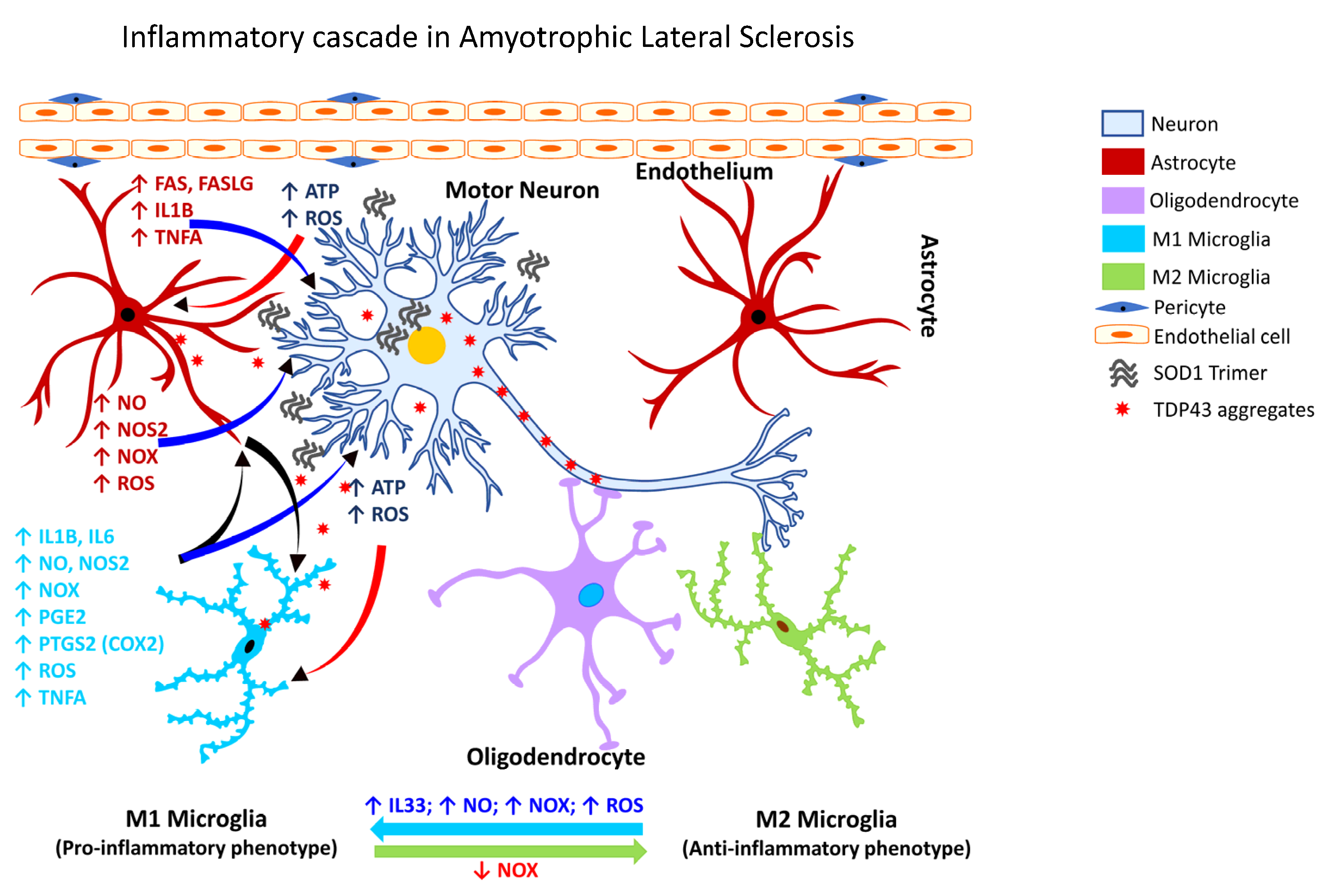
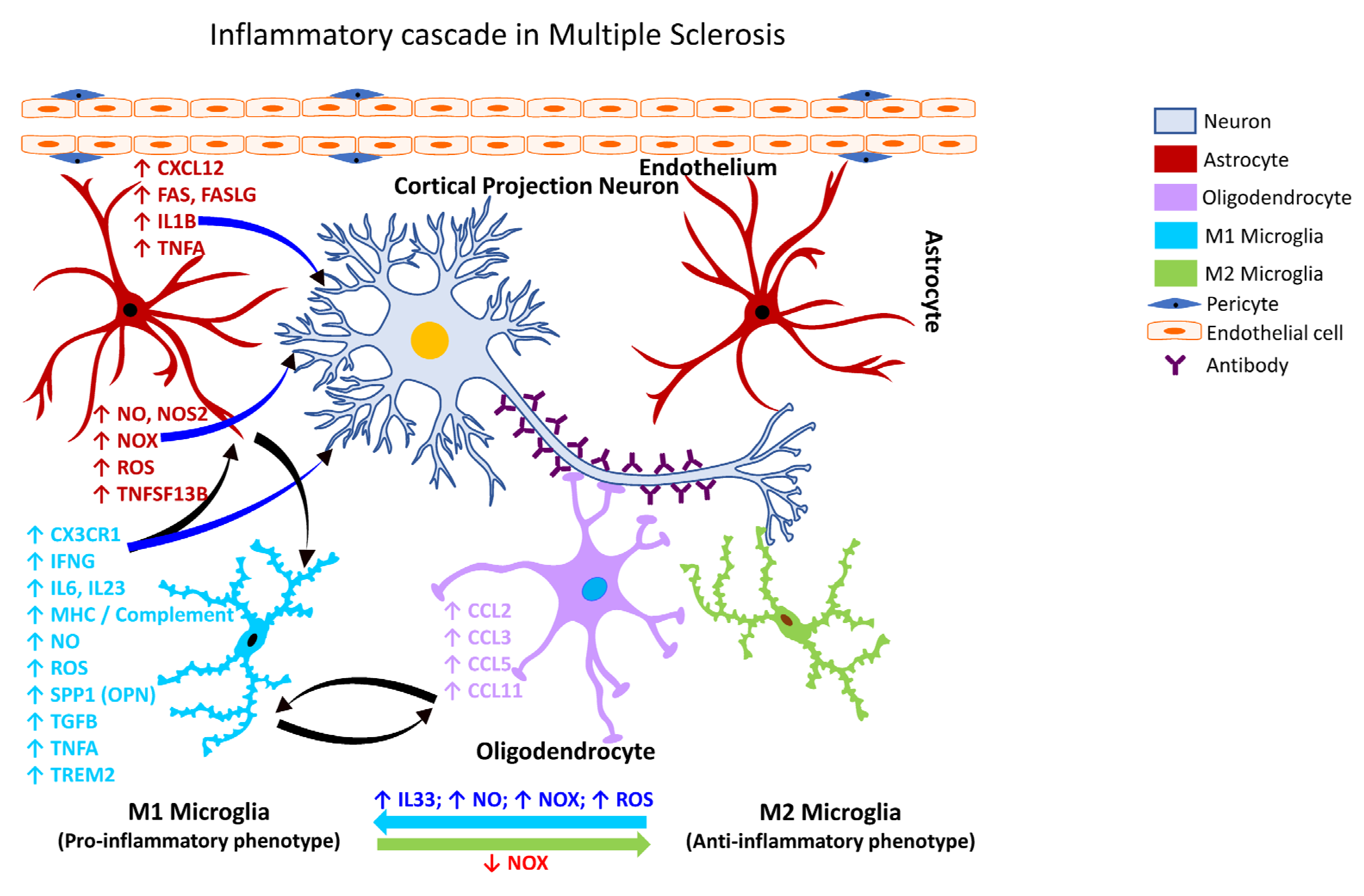
2.3.5. Gliosis in MS
References
- Varacallo, M.; Knoblauch, D.K. Occupational Injuries and Workers’ Compensation Management Strategies; StatPearls Publishing: Treasure Island, FL, USA, 2022.
- Mayo Clinic. Understanding Brain Injury, A Guide for Employers. MC1298rev0111, 2011, mc1298-2. Available online: https://biaia.org/ (accessed on 1 December 2022).
- Available online: https://injuryfacts.nsc.org/all-injuries/deaths-by-demographics/top-10-preventable-injuries/ (accessed on 1 December 2022).
- Pek, S.; Turner, N.; Tucker, S.; Kelloway, E.K.; Morrish, J. Injunctive safety norms, young worker risk-taking behaviors, and workplace injuries. Accid. Anal. Prev. 2017, 106, 202–210.
- NIOSH. Slip, Trip, and Fall Prevention for Health Care Workers. DHHS (NIOSH) Publication No. 2011-123. 2010. Available online: http://www.cdc.gov/niosh/docs/2011-123 (accessed on 1 December 2022).
- NIOSH. Preventing Slips, Trips, and Falls in Wholesale and Retail Trade Establishments DHHS (NIOSH) Publication No. 2013−100, 2013-100.pdf. 2012. Available online: https://www.cdc.gov/ (accessed on 1 December 2022).
- Anderson, V.P.; Mulhern, B. Focus on risk management. Don’t let costly slip and fall injuries trip you up. Chain Storage Age 2010, 86, 42–43.
- Terry, D.P.; Iverson, G.L.; Panenka, W.; Colantonio, A.; Silverberg, N.D. Workplace and non-workplace mild traumatic brain injuries in an outpatient clinic sample: A case-control study. PLoS ONE 2018, 13, e0198128.
- Postolache, T.T.; Wadhawan, A.; Can, A.; Lowry, C.A.; Woodbury, M.; Makkar, H.; Hoisington, A.J.; Scott, A.J.; Potocki, E.; Benros, M.E.; et al. Inflammation in Traumatic Brain Injury. J. Alzheimer’s Dis. 2020, 74, 1–28.
- Tiesman, H.; Konda, S.; Bell, J. The Epidemiology of Fatal Occupational Traumatic Brain Injury in the United States. Am. J. Prev. Med. 2011, 41, 61–67.
- Konda, S.; Tiesman, H.; Reichard, A. Nonfatal occupational traumatic brain injuries treated in U.S. hospital emergency departments: 1998–2007. Inj. Prev. 2015, 21, 115–120.
- Ogbodo, J.O.; Arazu, A.V.; Iguh, T.C.; Onwodi, N.J.; Ezike, T.C. Volatile organic compounds: A proinflammatory activator in autoimmune diseases. Front. Immunol. 2022, 13, 928379.
- Ruiz-Hernandez, A.; Kuo, C.C.; Rentero-Garrido, P.; Tang, W.Y.; Redon, J.; Ordovas, J.M.; Navas-Acien, A.; Tellez-Plaza, M. Environmental chemicals and DNA methylation in adults: A systematic review of the epidemiologic evidence. Clin. Epigenetics 2015, 7, 55.
- Zhang, Q.; Niu, Q.; Li, L.Y.; Yang, L.; Guo, X.L.; Huang, J.X.; Wang, L.P.; Liang, Y.X. Establishment of a poisoned animal model of toxic encephalopathy induced by 1,2-dichloroethane. Int. J. Immunopathol. Pharmacol. 2011, 24 (Suppl. S1), 79S–83S.
- Chen, S.; Zhang, Z.; Lin, H.; Chen, Z.; Wang, Z.; Wang, W. 1,2-Dichloroethane-induced toxic encephalopathy: A case series with morphological investigations. J. Neurol. Sci. 2015, 351, 36–40.
- Yang, J.; Wang, T.; Jin, X.; Wang, G.; Zhao, F.; Jin, Y. Roles of Crosstalk between Astrocytes and Microglia in Triggering Neuroinflammation and Brain Edema Formation in 1,2-Dichloroethane-Intoxicated Mice. Cells 2021, 10, 2647.
- Wang, T.; Sun, Q.; Yang, J.; Wang, G.; Zhao, F.; Chen, Y.; Jin, Y. Reactive astrocytes induced by 2-chloroethanol modulate microglia polarization through IL-1β, TNF-α, and iNOS upregulation. Food Chem. Toxicol. 2021, 157, 112550.
- Zhong, Y.; Liang, B.; Meng, H.; Ye, R.; Li, Z.; Du, J.; Wang, B.; Zhang, B.; Huang, Y.; Lin, X.; et al. 1,2-Dichloroethane induces cortex demyelination by depressing myelin basic protein via inhibiting aquaporin 4 in mice. Ecotoxicol. Environ. Saf. 2022, 231, 113180.
- Sriram, K.; O’Callaghan, J.P. Signaling Mechanisms Underlying Toxicant-Induced Gliosis. In The Role of Glia in Neurotoxicity, 2nd ed.; Aschner, M., Costa, L.G., Eds.; CRC Press: Boca Raton, FL, USA, 2005; pp. 141–171.
- O’Callaghan, J.P.; Sriram, K. Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Expert Opin. Drug Saf. 2005, 4, 433–442.
- Tjalkens, R.B.; Popichak, K.A.; Kirkley, K.A. Inflammatory Activation of Microglia and Astrocytes in Manganese Neurotoxicity. Adv. Neurobiol. 2017, 18, 159–181.
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42.
- Rosengren, L.E.; Aurell, A.; Kjellstrand, P.; Haglid, K.G. Astrogliosis in the cerebral cortex of gerbils after long-term exposure to 1,1,1-trichloroethane. Scand. J. Work Environ. Health 1985, 11, 447–455.
- Shi, J.; Ma, Y.; Zheng, M.; Ruan, Z.; Liu, J.; Tian, S.; Zhang, D.; He, X.; Li, G. Effect of sub-acute exposure to acrylamide on GABAergic neurons and astrocytes in weaning rat cerebellum. Toxicol. Ind. Health 2012, 28, 10–20.
- Garcia, S.J.; Seidler, F.J.; Qiao, D.; Slotkin, T.A. Chlorpyrifos targets developing glia: Effects on glial fibrillary acidic protein. Brain Res. Dev. Brain Res. 2002, 133, 151–161.
- Campos-Ordonez, T.; Zarate-Lopez, D.; Galvez-Contreras, A.Y.; Moy-Lopez, N.; Guzman-Muniz, J.; Gonzalez-Perez, O. Cyclohexane produces behavioral deficits associated with astrogliosis and microglial reactivity in the adult hippocampus mouse brain. Cell Mol. Neurobiol. 2015, 35, 503–512.
- Rosengren, L.E.; Kjellstrand, P.; Aurell, A.; Haglid, K.G. Irreversible effects of dichloromethane on the brain after long term exposure: A quantitative study of DNA and the glial cell marker proteins S-100 and GFA. Br. J. Ind. Med. 1986, 43, 291–299.
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; McKinney, W.; Jackson, M.C.; Cumpston, A.; Cumpston, J.L.; Cumpston, J.B.; Leonard, H.D.; Kashon, M.; et al. Biological effects of inhaled hydraulic fracturing sand dust VII. Neuroinflammation and altered synaptic protein expression. Toxicol. Appl. Pharmacol. 2020, 409, 115300.
- Olanow, C.W.; Good, P.F.; Shinotoh, H.; Hewitt, K.A.; Vingerhoets, F.; Snow, B.J.; Beal, M.F.; Calne, D.B.; Perl, D.P. Manganese intoxication in the rhesus monkey: A clinical, imaging, pathologic, and biochemical study. Neurology 1996, 46, 492–498.
- Vezér, T.; Kurunczi, A.; Náray, M.; Papp, A.; Nagymajtényi, L. Behavioral effects of subchronic inorganic manganese exposure in rats. Am. J. Ind. Med. 2007, 50, 841–852.
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; Goldsmith, W.T.; Jackson, M.; McKinney, W.; Frazer, D.G.; Robinson, V.A.; Castranova, V. Neurotoxicity following acute inhalation exposure to the oil dispersant COREXIT EC9500A. J. Toxicol. Environ. Health A 2011, 74, 1405–1418.
- Opris, R.V.; Toma, V.; Baciu, A.M.; Moldovan, R.; Dume, B.; Berghian-Sevastre, A.; Moldovan, B.; Clichici, S.; David, L.; Filip, G.A.; et al. Neurobehavioral and Ultrastructural Changes Induced by Phytosynthesized Silver-Nanoparticle Toxicity in an In Vivo Rat Model. Nanomaterials 2021, 12, 58.
- Fukui, K.; Utsumi, H.; Tamada, Y.; Nakajima, T.; Ibata, Y. Selective increase in astrocytic elements in the rat dentate gyrus after chronic toluene exposure studied by GFAP immunocytochemistry and electron microscopy. Neurosci. Lett. 1996, 203, 85–88.
- Gotohda, T.; Kuwada, A.; Morita, K.; Kubo, S.; Tokunaga, I. Elevation of steroid 5 alpha-reductase mRNA levels in rat cerebellum by toluene inhalation: Possible relation to GFAP expression. J. Toxicol. Sci. 2000, 25, 223–231.
- Svenson, D.W.; Davidson, C.J.; Thakur, C.; Bowen, S.E. Acute exposure to abuse-like concentrations of toluene induces inflammation in mouse lungs and brain. J. Appl. Toxicol. 2022, 42, 1168–1177.
- Kuhlmann, A.C.; Guilarte, T.R. Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J. Neurochem. 2000, 74, 1694–1704.
- Fiedorowicz, A.; Figiel, I.; Kamińska, B.; Zaremba, M.; Wilk, S.; Oderfeld-Nowak, B. Dentate granule neuron apoptosis and glia activation in murine hippocampus induced by trimethyltin exposure. Brain Res. 2001, 912, 116–127.
- Antonini, J.M.; Sriram, K.; Benkovic, S.A.; Roberts, J.R.; Stone, S.; Chen, B.T.; Schwegler-Berry, D.; Jefferson, A.M.; Billig, B.K.; Felton, C.M.; et al. Mild steel welding fume causes manganese accumulation and subtle neuroinflammatory changes but not overt neuronal damage in discrete brain regions of rats after short-term inhalation exposure. Neurotoxicology 2009, 30, 915–925.
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; Roberts, J.R.; Chapman, R.S.; Chen, B.T.; Soukup, J.M.; Ghio, A.J.; Antonini, J.M. Dopaminergic neurotoxicity following pulmonary exposure to manganese-containing welding fumes. Arch. Toxicol. 2010, 84, 521–540.
- Ajtai, B.M.; Kálmán, M. Glial fibrillary acidic protein expression but no glial demarcation follows the lesion in the molecular layer of cerebellum. Brain Res. 1998, 802, 285–288.
- Krum, J.M.; Phillips, T.M.; Rosenstein, J.M. Changes in astroglial GLT-1 expression after neural transplantation or stab wounds. Exp. Neurol. 2002, 174, 137–149.
- Isono, M.; Goda, M.; Kobayashi, H.; Wu, J.L. TGF-alpha overexpression induces astrocytic hypertrophy after cortical stab wound injury. Neurol. Res. 2003, 25, 546–550.
- Laskawi, R.; Rohlmann, A.; Landgrebe, M.; Wolff, J.R. Rapid astroglial reactions in the motor cortex of adult rats following peripheral facial nerve lesions. Eur. Arch. Otorhinolaryngol. 1997, 254, 81–85.
- Carbonell, W.S.; Mandell, J.W. Transient neuronal but persistent astroglial activation of ERK/MAP kinase after focal brain injury in mice. J. Neurotrauma 2003, 20, 327–336.
- Zhu, W.; Umegaki, H.; Shinkai, T.; Kurotani, S.; Suzuki, Y.; Endo, H.; Iguchi, A. Different glial reactions to hippocampal stab wounds in young adult and aged rats. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 117–122.
- Cheung, W.M.; Wang, C.K.; Kuo, J.S.; Lin, T.N. Changes in the level of glial fibrillary acidic protein (GFAP) after mild and severe focal cerebral ischemia. Chin. J. Physiol. 1999, 42, 227–235.
- Endoh, M.; Maiese, K.; Wagner, J. Expression of the inducible form of nitric oxide synthase by reactive astrocytes after transient global ischemia. Brain Res. 1994, 651, 92–100.
- Soltys, Z.; Janeczko, K.; Orzyłowska-Sliwińska, O.; Zaremba, M.; Januszewski, S.; Oderfeld-Nowak, B. Morphological transformations of cells immunopositive for GFAP, TrkA or p75 in the CA1 hippocampal area following transient global ischemia in the rat. A quantitative study. Brain Res. 2003, 987, 186–193.
- Butler, T.L.; Kassed, C.A.; Sanberg, P.R.; Willing, A.E.; Pennypacker, K.R. Neurodegeneration in the rat hippocampus and striatum after middle cerebral artery occlusion. Brain Res. 2002, 929, 252–260.
- Wang, H.; Zhang, Y.P.; Cai, J.; Shields, L.B.; Tuchek, C.A.; Shi, R.; Li, J.; Shields, C.B.; Xu, X.M. A Compact Blast-Induced Traumatic Brain Injury Model in Mice. J. Neuropathol. Exp. Neurol. 2016, 75, 183–196.
- Witcher, K.G.; Bray, C.E.; Chunchai, T.; Zhao, F.; O’Neil, S.M.; Gordillo, A.J.; Campbell, W.A.; McKim, D.B.; Liu, X.; Dziabis, J.E.; et al. Traumatic Brain Injury Causes Chronic Cortical Inflammation and Neuronal Dysfunction Mediated by Microglia. J. Neurosci. 2021, 41, 1597–1616.
- Ravula, A.R.; Rodriguez, J.; Younger, D.; Perumal, V.; Shao, N.; Rama Rao, K.V.; Pfister, B.; Chandra, N. Animal model of repeated low-level blast traumatic brain injury displays acute and chronic neurobehavioral and neuropathological changes. Exp. Neurol. 2022, 349, 113938.
- Panter, S.S.; McSwigan, J.D.; Sheppard, J.R.; Emory, C.R.; Frey, W.H., 2nd. Glial fibrillary acidic protein and Alzheimer’s disease. Neurochem. Res. 1985, 10, 1567–1576.
- Yamaguchi, H.; Morimatsu, M.; Hirai, S.; Takahashi, K. Alzheimer’s neurofibrillary tangles are penetrated by astroglial processes and appear eosinophilic in their final stages. Acta Neuropathol. 1987, 72, 214–217.
- Delacourte, A. General and dramatic glial reaction in Alzheimer brains. Neurology 1990, 40, 33–37.
- Vijayan, V.K.; Geddes, J.W.; Anderson, K.J.; Chang-Chui, H.; Ellis, W.G.; Cotman, C.W. Astrocyte hypertrophy in the Alzheimer’s disease hippocampal formation. Exp. Neurol. 1991, 112, 72–78.
- Cullen, K.M. Perivascular astrocytes within Alzheimer’s disease plaques. Neuroreport 1997, 8, 1961–1966.
- Kumar, A.; Koistinen, N.A.; Malarte, M.L.; Nennesmo, I.; Ingelsson, M.; Ghetti, B.; Lemoine, L.; Nordberg, A. Astroglial tracer BU99008 detects multiple binding sites in Alzheimer’s disease brain. Mol. Psychiatry 2021, 26, 5833–5847.
- Astillero-Lopez, V.; Gonzalez-Rodriguez, M.; Villar-Conde, S.; Flores-Cuadrado, A.; Martinez-Marcos, A.; Ubeda-Banon, I.; Saiz-Sanchez, D. Neurodegeneration and astrogliosis in the entorhinal cortex in Alzheimer’s disease: Stereological layer-specific assessment and proteomic analysis. Alzheimers Dement. 2022, 18, 2468–2480.
- Harpin, M.L.; Delaère, P.; Javoy-Agid, F.; Bock, E.; Jacque, C.; Delpech, B.; Villarroya, H.; Duyckaerts, C.; Hauw, J.J.; Baumann, N. Glial fibrillary acidic protein and beta A4 protein deposits in temporal lobe of aging brain and senile dementia of the Alzheimer type: Relation with the cognitive state and with quantitative studies of senile plaques and neurofibrillary tangles. J. Neurosci. Res. 1990, 27, 587–594.
- Troost, D.; Sillevis Smitt, P.A.; de Jong, J.M.; Swaab, D.F. Neurofilament and glial alterations in the cerebral cortex in amyotrophic lateral sclerosis. Acta Neuropathol. 1992, 84, 664–673.
- Aoki, T.; Kobayashi, K.; Isaki, K. Microglial and astrocytic change in brains of Creutzfeldt-Jakob disease: An immunocytochemical and quantitative study. Clin. Neuropathol. 1999, 18, 51–60.
- Zanusso, G.; Colaizzo, E.; Poleggi, A.; Masullo, C.; Romeo, R.; Ferrari, S.; Bongianni, M.; Fiorini, M.; Tiple, D.; Vaianella, L.; et al. Biochemical and Neuropathological Findings in a Creutzfeldt-Jakob Disease Patient with the Rare Val180Ile-129Val Haplotype in the Prion Protein Gene. Int. J. Mol. Sci. 2022, 23, 10210.
- Petzold, A.; Eikelenboom, M.J.; Gveric, D.; Keir, G.; Chapman, M.; Lazeron, R.H.; Cuzner, M.L.; Polman, C.H.; Uitdehaag, B.M.; Thompson, E.J.; et al. Markers for different glial cell responses in multiple sclerosis: Clinical and pathological correlations. Brain 2002, 125 Pt 7, 1462–1473.
- Nowacki, P.; Koziarska, D.; Masztalewicz, M. Microglia and astroglia proliferation within the normal appearing white matter in histologically active and inactive multiple sclerosis. Folia Neuropathol. 2019, 57, 249–257.
- Hishikawa, N.; Hashizume, Y.; Yoshida, M.; Sobue, G. Widespread occurrence of argyrophilic glial inclusions in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2001, 27, 362–372.
- Flores-Cuadrado, A.; Saiz-Sanchez, D.; Mohedano-Moriano, A.; Lamas-Cenjor, E.; Leon-Olmo, V.; Martinez-Marcos, A.; Ubeda-Bañon, I. Astrogliosis and sexually dimorphic neurodegeneration and microgliosis in the olfactory bulb in Parkinson’s disease. NPJ Parkinson’s Dis. 2021, 7, 11.
- Arnold, S.E.; Franz, B.R.; Trojanowski, J.Q.; Moberg, P.J.; Gur, R.E. Glial fibrillary acidic protein-immunoreactive astrocytosis in elderly patients with schizophrenia and dementia. Acta Neuropathol. 1996, 91, 269–277.
- Zhang, C.; Hou, L.; Yang, J.; Che, Y.; Sun, F.; Li, H.; Wang, Q. 2,5-Hexanedione induces dopaminergic neurodegeneration through integrin αMβ2/NADPH oxidase axis-mediated microglial activation. Cell Death Dis. 2018, 9, 60.
- Liu, Y.; Wang, Y.; Zhang, X.; Jiao, Y.; Duan, L.; Dai, L.; Yan, H. Chronic acrylamide exposure resulted in dopaminergic neuron loss, neuroinflammation and motor impairment in rats. Toxicol. Appl. Pharmacol. 2022, 451, 116190.
- Platt, B.; Fiddler, G.; Riedel, G.; Henderson, Z. Aluminium toxicity in the rat brain: Histochemical and immunocytochemical evidence. Brain Res. Bull. 2001, 55, 257–267.
- Singh, A.K.; Tiwari, M.N.; Dixit, A.; Upadhyay, G.; Patel, D.K.; Singh, D.; Prakash, O.; Singh, M.P. Nigrostriatal proteomics of cypermethrin-induced dopaminergic neurodegeneration: Microglial activation-dependent and -independent regulations. Toxicol. Sci. 2011, 122, 526–538.
- Hubbs, A.F.; Fluharty, K.L.; Edwards, R.J.; Barnabei, J.L.; Grantham, J.T.; Palmer, S.M.; Kelly, F.; Sargent, L.M.; Reynolds, S.H.; Mercer, R.R.; et al. Accumulation of Ubiquitin and Sequestosome-1 Implicate Protein Damage in Diacetyl-Induced Cytotoxicity. Am. J. Pathol. 2016, 186, 2887–2908.
- Binukumar, B.K.; Bal, A.; Gill, K.D. Chronic dichlorvos exposure: Microglial activation, proinflammatory cytokines and damage to nigrostriatal dopaminergic system. NeuroMolecular Med. 2011, 13, 251–265.
- Levesque, S.; Taetzsch, T.; Lull, M.E.; Kodavanti, U.; Stadler, K.; Wagner, A.; Johnson, J.A.; Duke, L.; Kodavanti, P.; Surace, M.J.; et al. Diesel exhaust activates and primes microglia: Air pollution, neuroinflammation, and regulation of dopaminergic neurotoxicity. Environ. Health Perspect. 2011, 119, 1149–1155.
- Zucconi, G.G.; Laurenzi, M.A.; Semprevivo, M.; Torni, F.; Lindgren, J.A.; Marinucci, E. Microglia activation and cell death in response to diethyl-dithiocarbamate acute administration. J. Comp. Neurol. 2002, 446, 135–150.
- Wang, Y.; Wang, B.; Zhu, M.T.; Li, M.; Wang, H.J.; Wang, M.; Ouyang, H.; Chai, Z.F.; Feng, W.Y.; Zhao, Y.L. Microglial activation, recruitment and phagocytosis as linked phenomena in ferric oxide nanoparticle exposure. Toxicol. Lett. 2011, 205, 26–37.
- Zhao, F.; Cai, T.; Liu, M.; Zheng, G.; Luo, W.; Chen, J. Manganese induces dopaminergic neurodegeneration via microglial activation in a rat model of manganism. Toxicol. Sci. 2009, 107, 156–164.
- Gonzalez-Cuyar, L.F.; Nelson, G.; Criswell, S.R.; Ho, P.; Lonzanida, J.A.; Checkoway, H.; Seixas, N.; Gelman, B.B.; Evanoff, B.A.; Murray, J.; et al. Quantitative neuropathology associated with chronic manganese exposure in South African mine workers. Neurotoxicology 2014, 45, 260–266.
- Hou, L.; Zhang, C.; Wang, K.; Liu, X.; Wang, H.; Che, Y.; Sun, F.; Zhou, X.; Zhao, X.; Wang, Q. Paraquat and maneb co-exposure induces noradrenergic locus coeruleus neurodegeneration through NADPH oxidase-mediated microglial activation. Toxicology 2017, 380, 1–10.
- Sherer, T.B.; Betarbet, R.; Kim, J.H.; Greenamyre, J.T. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci. Lett. 2003, 341, 87–90.
- Zhang, D.; Li, S.; Hou, L.; Jing, L.; Ruan, Z.; Peng, B.; Zhang, X.; Hong, J.S.; Zhao, J.; Wang, Q. Microglial activation contributes to cognitive impairments in rotenone-induced mouse Parkinson’s disease model. J. Neuroinflamm. 2021, 18, 4.
- Patchin, E.S.; Anderson, D.S.; Silva, R.M.; Uyeminami, D.L.; Scott, G.M.; Guo, T.; Van Winkle, L.S.; Pinkerton, K.E. Size-Dependent Deposition, Translocation, and Microglial Activation of Inhaled Silver Nanoparticles in the Rodent Nose and Brain. Environ. Health Perspect. 2016, 124, 1870–1875.
- Huynh, H.; Upadhyay, P.; Lopez, C.H.; Miyashiro, M.K.; Van Winkle, L.S.; Thomasy, S.M.; Pinkerton, K.E. Inhalation of Silver Silicate Nanoparticles Leads to Transient and Differential Microglial Activation in the Rodent Olfactory Bulb. Toxicol. Pathol. 2022, 50, 763–775.
- Koczyk, D.; Oderfeld-Nowak, B. Long-term microglial and astroglial activation in the hippocampus of trimethyltin-intoxicated rat: Stimulation of NGF and TrkA immunoreactivities in astroglia but not in microglia. Int. J. Dev. Neurosci. 2000, 18, 591–606.
- Sriram, K.; Jefferson, A.M.; Lin, G.X.; Afshari, A.; Zeidler-Erdely, P.C.; Meighan, T.G.; McKinney, W.; Jackson, M.; Cumpston, A.; Cumpston, J.L.; et al. Neurotoxicity following acute inhalation of aerosols generated during resistance spot weld-bonding of carbon steel. Inhal. Toxicol. 2014, 26, 720–732.
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; Stone, S.; Afshari, A.; Keane, M.J.; McKinney, W.; Jackson, M.; Chen, B.T.; Schwegler-Berry, D.; et al. Modifying welding process parameters can reduce the neurotoxic potential of manganese-containing welding fumes. Toxicology 2015, 328, 168–178.
- Katchanov, J.; Waeber, C.; Gertz, K.; Gietz, A.; Winter, B.; Brück, W.; Dirnagl, U.; Veh, R.W.; Endres, M. Selective neuronal vulnerability following mild focal brain ischemia in the mouse. Brain Pathol. 2003, 13, 452–464.
- Huh, Y.; Jung, J.W.; Park, C.; Ryu, J.R.; Shin, C.Y.; Kim, W.K.; Ryu, J.H. Microglial activation and tyrosine hydroxylase immunoreactivity in the substantia nigral region following transient focal ischemia in rats. Neurosci. Lett. 2003, 349, 63–67.
- Lin, B.; Ginsberg, M.D.; Busto, R.; Dietrich, W.D. Sequential analysis of subacute and chronic neuronal, astrocytic and microglial alterations after transient global ischemia in rats. Acta Neuropathol. 1998, 95, 511–523.
- Dihné, M.; Block, F. Focal ischemia induces transient expression of IL-6 in the substantia nigra pars reticulata. Brain Res. 2001, 889, 165–173.
- DeKosky, S.T.; Styren, S.D.; O’Malley, M.E.; Goss, J.R.; Kochanek, P.; Marion, D.; Evans, C.H.; Robbins, P.D. Interleukin-1 receptor antagonist suppresses neurotrophin response in injured rat brain. Ann. Neurol. 1996, 39, 123–127.
- Scott, G.; Zetterberg, H.; Jolly, A.; Cole, J.H.; De Simoni, S.; Jenkins, P.O.; Feeney, C.; Owen, D.R.; Lingford-Hughes, A.; Howes, O.; et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain 2018, 141, 459–471.
- Wierzba-Bobrowicz, T.; Gwiazda, E.; Kosno-Kruszewska, E.; Lewandowska, E.; Lechowicz, W.; Bertrand, E.; Szpak, G.M.; Schmidt-Sidor, B. Morphological analysis of active microglia--rod and ramified microglia in human brains affected by some neurological diseases (SSPE, Alzheimer’s disease and Wilson’s disease). Folia Neuropathol. 2002, 40, 125–131.
- Versijpt, J.J.; Dumont, F.; Van Laere, K.J.; Decoo, D.; Santens, P.; Audenaert, K.; Achten, E.; Slegers, G.; Dierckx, R.A.; Korf, J. Assessment of neuroinflammation and microglial activation in Alzheimer’s disease with radiolabelled PK11195 and single photon emission computed tomography. A pilot study. Eur. Neurol. 2003, 50, 39–47.
- Walker, D.G.; Tang, T.M.; Lue, L.F. Studies on Colony Stimulating Factor Receptor-1 and Ligands Colony Stimulating Factor-1 and Interleukin-34 in Alzheimer’s Disease Brains and Human Microglia. Front. Aging Neurosci. 2017, 9, 244.
- Singh-Bains, M.K.; Linke, V.; Austria, M.D.R.; Tan, A.Y.S.; Scotter, E.L.; Mehrabi, N.F.; Faull, R.L.M.; Dragunow, M. Altered microglia and neurovasculature in the Alzheimer’s disease cerebellum. Neurobiol. Dis. 2019, 132, 104589.
- Fixemer, S.; Ameli, C.; Hammer, G.; Salamanca, L.; Uriarte Huarte, O.; Schwartz, C.; Gérardy, J.J.; Mechawar, N.; Skupin, A.; Mittelbronn, M.; et al. Microglia phenotypes are associated with subregional patterns of concomitant tau, amyloid-β and α-synuclein pathologies in the hippocampus of patients with Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. Commun. 2022, 10, 36.
- Fracassi, A.; Marcatti, M.; Tumurbaatar, B.; Woltjer, R.; Moreno, S.; Taglialatela, G. TREM2-induced activation of microglia contributes to synaptic integrity in cognitively intact aged individuals with Alzheimer’s neuropathology. Brain Pathol. 2022, 33, e13108.
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467, Erratum in Lancet 2001, 358, 766.
- Wilson, C.M.; Grace, G.M.; Munoz, D.G.; He, B.P.; Strong, M.J. Cognitive impairment in sporadic ALS: A pathologic continuum underlying a multisystem disorder. Neurology 2001, 57, 651–657.
- Brettschneider, J.; Libon, D.J.; Toledo, J.B.; Xie, S.X.; McCluskey, L.; Elman, L.; Geser, F.; Lee, V.M.; Grossman, M.; Trojanowski, J.Q. Microglial activation and TDP-43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 123, 395–407.
- Sugiyama, M.; Takao, M.; Hatsuta, H.; Funabe, S.; Ito, S.; Obi, T.; Tanaka, F.; Kuroiwa, Y.; Murayama, S. Increased number of astrocytes and macrophages/microglial cells in the corpus callosum in amyotrophic lateral sclerosis. Neuropathology 2013, 33, 591–599.
- Gray, F.; Chrétien, F.; Adle-Biassette, H.; Dorandeu, A.; Ereau, T.; Delisle, M.B.; Kopp, N.; Ironside, J.W.; Vital, C. Neuronal apoptosis in Creutzfeldt-Jakob disease. J. Neuropathol. Exp. Neurol. 1999, 58, 321–328.
- Schwarz, S.C.; Seufferlein, T.; Liptay, S.; Schmid, R.M.; Kasischke, K.; Foster, O.J.; Daniel, S.; Schwarz, J. Microglial activation in multiple system atrophy: A potential role for NF-kappaB/rel proteins. Neuroreport 1998, 9, 3029–3032.
- Banati, R.B.; Daniel, S.E.; Blunt, S.B. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov. Disord. 1998, 13, 221–227.
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The role of glial reaction and inflammation in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 214–228.
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248.
- Harry, G.J.; Schmitt, T.J.; Gong, Z.; Brown, H.; Zawia, N.; Evans, H.L. Lead-induced alterations of glial fibrillary acidic protein (GFAP) in the developing rat brain. Toxicol. Appl. Pharmacol. 1996, 139, 84–93.
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024.
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934.
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832.
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315.
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218.
- Murakami, M.; Hirano, T. The molecular mechanisms of chronic inflammation development. Front. Immunol. 2012, 3, 323.
- Yeh, H.; Ikezu, T. Transcriptional and Epigenetic Regulation of Microglia in Health and Disease. Trends Mol. Med. 2019, 25, 96–111.
- Cramer, T.; Gill, R.; Thirouin, Z.S.; Vaas, M.; Sampath, S.; Martineau, F.; Noya, S.B.; Panzanelli, P.; Sudharshan, T.J.J.; Colameo, D.; et al. Cross-talk between GABAergic postsynapse and microglia regulate synapse loss after brain ischemia. Sci. Adv. 2022, 8, eabj0112.
- Van Everbroeck, B.; Dobbeleir, I.; De Waele, M.; De Leenheir, E.; Lübke, U.; Martin, J.J.; Cras, P. Extracellular protein deposition correlates with glial activation and oxidative stress in Creutzfeldt-Jakob and Alzheimer’s disease. Acta Neuropathol. 2004, 108, 194–200.
- Neidert, N.; von Ehr, A.; Zöller, T.; Spittau, B. Microglia-Specific Expression of Olfml3 Is Directly Regulated by Transforming Growth Factor β1-Induced Smad2 Signaling. Front. Immunol. 2018, 9, 1728.
- Sears, J.M.; Graves, J.M.; Blanar, L.; Bowman, S.M. Case identification of work-related traumatic brain injury using the occupational injury and illness classification system. J. Occup. Environ. Med. 2013, 55, 507–513.
- Dorsett, C.R.; McGuire, J.L.; DePasquale, E.A.; Gardner, A.E.; Floyd, C.L.; McCullumsmith, R.E. Glutamate Neurotransmission in Rodent Models of Traumatic Brain Injury. J. Neurotrauma 2017, 34, 263–272.
- Chen, C.; Hou, J.; Lu, J.; Zhu, Z.; Yang, Y.; Peng, W.; Pi, R. A novel simple traumatic brain injury mouse model. Chin. Neurosurg. J. 2022, 8, 8.
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265.
- Faul, M.; Wald, M.M.; Xu, L.; Coronado, V.G. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths 2002–2006; US Department of Health and Human Services, CDC: Atlanta, GA, USA, 2010.
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 572, Erratum in Nat. Rev. Neurol. 2017, 13, 171–191.
- Peterson, A.B.; Zhou, H.; Thomas, K.E. Disparities in traumatic brain injury-related deaths-United States, 2020. J. Saf. Res. 2022, 83, 419–426.
- De Crescenzo, L.A.; Gabella, B.A.; Johnson, J. Interrupted time series design to evaluate ICD-9-CM to ICD-10-CM coding changes on trends in Colorado emergency department visits related to traumatic brain injury. Inj. Epidemiol. 2021, 8, 15.
- Anderson, T.; Heitger, M.; Macleod, A.D. Concussion and mild head injury. Pract. Neurol. 2006, 6, 342–357.
- Available online: https://www.cdc.gov/headsup/basics/concussion_whatis.html (accessed on 1 December 2022).
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707.
- Anthonymuthu, T.S.; Kenny, E.M.; Bayır, H. Therapies targeting lipid peroxidation in traumatic brain injury. Brain Res. 2016, 1640, 57–76.
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208.
- Tyburski, A.L.; Cheng, L.; Assari, S.; Darvish, K.; Elliott, M.B. Frequent mild head injury promotes trigeminal sensitivity concomitant with microglial proliferation, astrocytosis, and increased neuropeptide levels in the trigeminal pain system. J. Headache Pain 2017, 18, 16.
- Verboon, L.N.; Patel, H.C.; Greenhalgh, A.D. The Immune System’s Role in the Consequences of Mild Traumatic Brain Injury (Concussion). Front. Immunol. 2021, 12, 620698.
- Cherry, J.D.; Tripodis, Y.; Alvarez, V.E.; Huber, B.; Kiernan, P.T.; Daneshvar, D.H.; Mez, J.; Montenigro, P.H.; Solomon, T.M.; Alosco, M.L.; et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol. Commun. 2016, 4, 112.
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228.
- Madathil, S.K.; Wilfred, B.S.; Urankar, S.E.; Yang, W.; Leung, L.Y.; Gilsdorf, J.S.; Shear, D.A. Early Microglial Activation Following Closed-Head Concussive Injury Is Dominated by Pro-Inflammatory M-1 Type. Front. Neurol. 2018, 9, 964.
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647.
- Sofroniew, M.V. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 2014, 20, 160–172.
- Witcher, K.G.; Eiferman, D.S.; Godbout, J.P. Priming the inflammatory pump of the CNS after traumatic brain injury. Trends Neurosci. 2015, 38, 609–620.
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell. 2018, 34, 863–867, Erratum in Cancer Cell. 2018, 33, 75–90.e7.
- Available online: https://ntp.niehs.nih.gov/whatwestudy/tox21/history/index.html (accessed on 1 December 2022).
- Thomas, R.S.; Paules, R.S.; Simeonov, A.; Fitzpatrick, S.C.; Crofton, K.M.; Casey, W.M.; Mendrick, D.L. The US Federal Tox21 Program: A strategic and operational plan for continued leadership. ALTEX 2018, 35, 163–168.
- Richard, A.M.; Huang, R.; Waidyanatha, S.; Shinn, P.; Collins, B.J.; Thillainadarajah, I.; Grulke, C.M.; Williams, A.J.; Lougee, R.R.; Judson, R.S.; et al. The Tox21 10K Compound Library: Collaborative Chemistry Advancing Toxicology. Chem. Res. Toxicol. 2021, 34, 189–216.
- Pezzoli, G.; Strada, O.; Silani, V.; Zecchinelli, A.; Perbellini, L.; Javoy-Agid, F.; Ghidoni, P.; Motti, E.D.; Masini, T.; Scarlato, G.; et al. Clinical and pathological features in hydrocarbon-induced parkinsonism. Ann. Neurol. 1996, 40, 922–925.
- Pezzoli, G.; Canesi, M.; Antonini, A.; Righini, A.; Perbellini, L.; Barichella, M.; Mariani, C.B.; Tenconi, F.; Tesei, S.; Zecchinelli, A.; et al. Hydrocarbon exposure and Parkinson’s disease. Neurology 2000, 55, 667–673.
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197.
- Chen, W.C.; Chang, L.H.; Huang, S.S.; Huang, Y.J.; Chih, C.L.; Kuo, H.C.; Lee, Y.H.; Lee, I.H. Aryl hydrocarbon receptor modulates stroke-induced astrogliosis and neurogenesis in the adult mouse brain. J. Neuroinflamm. 2019, 16, 187.
- Ramos-García, N.A.; Orozco-Ibarra, M.; Estudillo, E.; Elizondo, G.; Gómez Apo, E.; Chávez Macías, L.G.; Sosa-Ortiz, A.L.; Torres-Ramos, M.A. Aryl Hydrocarbon Receptor in Post-Mortem Hippocampus and in Serum from Young, Elder, and Alzheimer’s Patients. Int. J. Mol. Sci. 2020, 21, 1983.
- Cho, J.; Sohn, J.; Noh, J.; Jang, H.; Kim, W.; Cho, S.K.; Seo, H.; Seo, G.; Lee, S.K.; Noh, Y.; et al. Association between exposure to polycyclic aromatic hydrocarbons and brain cortical thinning: The Environmental Pollution-Induced Neurological EFfects (EPINEF) study. Sci. Total Environ. 2020, 737, 140097.
- Kim, Y.T.; Kim, W.; Bae, M.J.; Choi, J.E.; Kim, M.J.; Oh, S.S.; Park, K.S.; Park, S.; Lee, S.K.; Koh, S.B.; et al. The effect of polycyclic aromatic hydrocarbons on changes in the brain structure of firefighters: An analysis using data from the Firefighters Research on Enhancement of Safety & Health study. Sci. Total Environ. 2022, 816, 151655.
- Macchi, Z.A.; Carlisle, T.C.; Filley, C.M. Prognosis in substance abuse-related acute toxic leukoencephalopathy: A scoping review. J. Neurol. Sci. 2022, 442, 120420.
- Naskali, L.; Oksanen, H.; Tähti, H. Astrocytes as targets for CNS effects of organic solvents in vitro. Neurotoxicology 1994, 15, 609–612.
- Yamaguchi, H.; Kidachi, Y.; Ryoyama, K. Toluene at environmentally relevant low levels disrupts differentiation of astrocyte precursor cells. Arch. Environ. Health 2002, 57, 232–238.
- Riegel, A.C.; Zapata, A.; Shippenberg, T.S.; French, E.D. The abused inhalant toluene increases dopamine release in the nucleus accumbens by directly stimulating ventral tegmental area neurons. Neuropsychopharmacology 2007, 32, 1558–1569.
- Beckley, J.T.; Woodward, J.J. The abused inhalant toluene differentially modulates excitatory and inhibitory synaptic transmission in deep-layer neurons of the medial prefrontal cortex. Neuropsychopharmacology 2011, 36, 1531–1542.
- Chan, M.H.; Tang, Y.C.; Chien, T.H.; Chen, H.H. Toluene exposure during the brain growth spurt reduced behavioral responses to nicotine in young adult rats: A potential role for nicotinic acetylcholine receptors in fetal solvent syndrome. Toxicol. Sci. 2008, 101, 286–293.
- Gotohda, T.; Tokunaga, I.; Kubo, S.; Kitamura, O.; Ishigami, A. Toluene inhalation induces glial cell line-derived neurotrophic factor, transforming growth factor and tumor necrosis factor in rat cerebellum. Leg. Med. 2002, 4, 21–28.
- Montes, S.; Yee-Rios, Y.; Páez-Martínez, N. Environmental enrichment restores oxidative balance in animals chronically exposed to toluene: Comparison with melatonin. Brain Res. Bull. 2019, 144, 58–67.
- Zheng, W.; Monnot, A.D. Regulation of brain iron and copper homeostasis by brain barrier systems: Implication in neurodegenerative diseases. Pharmacol. Ther. 2012, 133, 177–188.
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 2011, 118, 939–957.
- Li, W.J.; Jiang, H.; Song, N.; Xie, J.X. Dose- and time-dependent alpha-synuclein aggregation induced by ferric iron in SK-N-SH cells. Neurosci. Bull. 2010, 26, 205–210.
- Kenkhuis, B.; Somarakis, A.; de Haan, L.; Dzyubachyk, O.; IJsselsteijn, M.E.; de Miranda, N.F.C.C.; Lelieveldt, B.P.F.; Dijkstra, J.; van Roon-Mom, W.M.C.; Höllt, T.; et al. Iron loading is a prominent feature of activated microglia in Alzheimer’s disease patients. Acta Neuropathol. Commun. 2021, 9, 27.
- Laabbar, W.; Abbaoui, A.; Elgot, A.; Mokni, M.; Amri, M.; Masmoudi-Kouki, O.; Gamrani, H. Aluminum induced oxidative stress, astrogliosis and cell death in rat astrocytes, is prevented by curcumin. J. Chem. Neuroanat. 2021, 112, 101915.
- Walton, J.R.; Wang, M.X. APP expression, distribution and accumulation are altered by aluminum in a rodent model for Alzheimer’s disease. J. Inorg. Biochem. 2009, 103, 1548–1554.
- Bhattacharjee, S.; Zhao, Y.; Hill, J.M.; Percy, M.E.; Lukiw, W.J. Aluminum and its potential contribution to Alzheimer’s disease (AD). Front. Aging Neurosci. 2014, 6, 62.
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Hill, J.; Dua, P.; Lukiw, W.J. Regulation of neurotropic signaling by the inducible, NF-kB-sensitive miRNA-125b in Alzheimer’s disease (AD) and in primary human neuronal-glial (HNG) cells. Mol. Neurobiol. 2014, 50, 97–106.
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell Neurosci. 2015, 9, 124.
- Rodier, J. Manganese poisoning in Moroccan miners. Br. J. Ind. Med. 1955, 12, 21–35.
- Donaldson, J. The physiopathologic significance of manganese in brain: Its relation to schizophrenia and neurodegenerative disorders. Neurotoxicology. 1987, 8, 451–462.
- Roels, H.; Lauwerys, R.; Buchet, J.P.; Genet, P.; Sarhan, M.J.; Hanotiau, I.; de Fays, M.; Bernard, A.; Stanescu, D. Epidemiological survey among workers exposed to manganese: Effects on lung, central nervous system, and some biological indices. Am. J. Ind. Med. 1987, 11, 307–327, Erratum in Am. J. Ind. Med. 1987, 12, 119–120.
- Mergler, D.; Huel, G.; Bowler, R.; Iregren, A.; Bélanger, S.; Baldwin, M.; Tardif, R.; Smargiassi, A.; Martin, L. Nervous system dysfunction among workers with long-term exposure to manganese. Environ. Res. 1994, 64, 151–180.
- Mergler, D.; Baldwin, M. Early manifestations of manganese neurotoxicity in humans: An update. Environ. Res. 1997, 73, 92–100.
- Bader, M.; Dietz, M.C.; Ihrig, A.; Triebig, G. Biomonitoring of manganese in blood, urine and axillary hair following low-dose exposure during the manufacture of dry cell batteries. Int. Arch. Occup. Environ. Health 1999, 72, 521–527.
- Lucchini, R.G.; Albini, E.; Benedetti, L.; Borghesi, S.; Coccaglio, R.; Malara, E.C.; Parrinello, G.; Garattini, S.; Resola, S.; Alessio, L. High prevalence of Parkinsonian disorders associated to manganese exposure in the vicinities of ferroalloy industries. Am. J. Ind. Med. 2007, 50, 788–800.
- Bowler, R.M.; Nakagawa, S.; Drezgic, M.; Roels, H.A.; Park, R.M.; Diamond, E.; Mergler, D.; Bouchard, M.; Bowler, R.P.; Koller, W. Sequelae of fume exposure in confined space welding: A neurological and neuropsychological case series. Neurotoxicology 2007, 28, 298–311.
- Bowler, R.M.; Roels, H.A.; Nakagawa, S.; Drezgic, M.; Diamond, E.; Park, R.; Koller, W.; Bowler, R.P.; Mergler, D.; Bouchard, M.; et al. Dose-effect relationships between manganese exposure and neurological, neuropsychological and pulmonary function in confined space bridge welders. Occup. Environ. Med. 2007, 64, 167–177.
- Guilarte, T.R. Manganese neurotoxicity: New perspectives from behavioral, neuroimaging, and neuropathological studies in humans and non-human primates. Front. Aging Neurosci. 2013, 5, 23.
- Park, R.M. Neurobehavioral deficits and parkinsonism in occupations with manganese exposure: A review of methodological issues in the epidemiological literature. Saf. Health Work 2013, 4, 123–135.
- Verina, T.; Schneider, J.S.; Guilarte, T.R. Manganese exposure induces α-synuclein aggregation in the frontal cortex of non-human primates. Toxicol. Lett. 2013, 217, 177–183.
- Aschner, M.; Aschner, J.L. Manganese transport across the blood-brain barrier: Relationship to iron homeostasis. Brain Res. Bull. 1990, 24, 857–860.
- Aschner, M.; Gannon, M. Manganese (Mn) transport across the rat blood-brain barrier: Saturable and transferrin-dependent transport mechanisms. Brain Res. Bull. 1994, 33, 345–349.
- Crossgrove, J.S.; Allen, D.D.; Bukaveckas, B.L.; Rhineheimer, S.S.; Yokel, R.A. Manganese distribution across the blood-brain barrier. I. Evidence for carrier-mediated influx of managanese citrate as well as manganese and manganese transferrin. Neurotoxicology 2003, 24, 3–13.
- Fitsanakis, V.A.; Piccola, G.; Marreilha dos Santos, A.P.; Aschner, J.L.; Aschner, M. Putative proteins involved in manganese transport across the blood-brain barrier. Hum. Exp. Toxicol. 2007, 26, 295–302.
- Conrad, M.E.; Umbreit, J.N.; Moore, E.G.; Hainsworth, L.N.; Porubcin, M.; Simovich, M.J.; Nakada, M.T.; Dolan, K.; Garrick, M.D. Separate pathways for cellular uptake of ferric and ferrous iron. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G767–G774.
- Crossgrove, J.S.; Yokel, R.A. Manganese distribution across the blood-brain barrier. IV. Evidence for brain influx through store-operated calcium channels. Neurotoxicology 2005, 26, 297–307.
- Finney, L.A.; O’Halloran, T.V. Transition metal speciation in the cell: Insights from the chemistry of metal ion receptors. Science 2003, 300, 931–936.
- Thompson, K.H.; Orvig, C. Boon and bane of metal ions in medicine. Science 2003, 300, 936–939.
- Thomassen, Y.; Ellingsen, D.G.; Hetland, S.; Sand, G. Chemical speciation and sequential extraction of Mn in workroom aerosols: Analytical methodology and results from a field study in Mn alloy plants. J. Environ. Monit. 2001, 3, 555–559.
- Reaney, S.H.; Bench, G.; Smith, D.R. Brain accumulation and toxicity of Mn(II) and Mn(III) exposures. Toxicol. Sci. 2006, 93, 114–124.
- Hunter, D.D.; Undem, B.J. Identification and substance P content of vagal afferent neurons innervating the epithelium of the guinea pig trachea. Am. J. Respir. Crit. Care Med. 1999, 159, 1943–1948.
- Oberdörster, G.; Sharp, Z.; Atudorei, V.; Elder, A.; Gelein, R.; Lunts, A.; Kreyling, W.; Cox, C. Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. J. Toxicol. Environ. Health A 2002, 65, 1531–1543.
- Oberdörster, G.; Sharp, Z.; Atudorei, V.; Elder, A.; Gelein, R.; Kreyling, W.; Cox, C. Translocation of inhaled ultrafine particles to the brain. Inhal. Toxicol. 2004, 16, 437–445.
- Elder, A.; Gelein, R.; Silva, V.; Feikert, T.; Opanashuk, L.; Carter, J.; Potter, R.; Maynard, A.; Ito, Y.; Finkelstein, J.; et al. Translocation of inhaled ultrafine manganese oxide particles to the central nervous system. Environ Health Perspect. 2006, 114, 1172–1178.
- Chandra, S.V.; Shukla, G.S.; Srivastava, R.S.; Singh, H.; Gupta, V.P. An exploratory study of manganese exposure to welders. Clin. Toxicol. 1981, 18, 407–416.
- Sjogren, U.; Hagglund, B.; Sundqvist, G.; Wing, K. Factors affecting the long-term results of endodontic treatment. J. Endod. 1990, 16, 498–504.
- Racette, B.A.; McGee-Minnich, L.; Moerlein, S.M.; Mink, J.W.; Videen, T.O.; Perlmutter, J.S. Welding-related parkinsonism: Clinical features, treatment, and pathophysiology. Neurology 2001, 56, 8–13.
- Bowler, R.M.; Gysens, S.; Diamond, E.; Booty, A.; Hartney, C.; Roels, H.A. Neuropsychological sequelae of exposure to welding fumes in a group of occupationally exposed men. Int. J. Hyg. Environ. Health 2003, 206, 517–529.
- Racette, B.A.; Tabbal, S.D.; Jennings, D.; Good, L.; Perlmutter, J.S.; Evanoff, B. Prevalence of parkinsonism and relationship to exposure in a large sample of Alabama welders. Neurology 2005, 64, 230–235.
- Josephs, K.A.; Ahlskog, J.E.; Klos, K.J.; Kumar, N.; Fealey, R.D.; Trenerry, M.R.; Cowl, C.T. Neurologic manifestations in welders with pallidal MRI T1 hyperintensity. Neurology 2005, 64, 2033–2039.
- Park, R.M.; Schulte, P.A.; Bowman, J.D.; Walker, J.T.; Bondy, S.C.; Yost, M.G.; Touchstone, J.A.; Dosemeci, M. Potential occupational risks for neurodegenerative diseases. Am. J. Ind. Med. 2005, 48, 63–77.
- Park, R.M.; Bowler, R.M.; Eggerth, D.E.; Diamond, E.; Spencer, K.J.; Smith, D.; Gwiazda, R. Issues in neurological risk assessment for occupational exposures: The Bay Bridge welders. Neurotoxicology 2006, 27, 373–384.
- Racette, B.A.; Searles Nielsen, S.; Criswell, S.R.; Sheppard, L.; Seixas, N.; Warden, M.N.; Checkoway, H. Dose-dependent progression of parkinsonism in manganese-exposed welders. Neurology 2017, 88, 344–351.
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; Roberts, J.R.; Wirth, O.; Hayashi, Y.; Krajnak, K.M.; Soukup, J.M.; Ghio, A.J.; Reynolds, S.H.; et al. Mitochondrial dysfunction and loss of Parkinson’s disease-linked proteins contribute to neurotoxicity of manganese-containing welding fumes. FASEB J. 2010, 24, 4989–5002.
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; Roberts, J.R.; Andrews, R.N.; Kashon, M.L.; Antonini, J.M. Manganese accumulation in nail clippings as a biomarker of welding fume exposure and neurotoxicity. Toxicology 2012, 291, 73–82.
- Ze, Y.; Sheng, L.; Zhao, X.; Hong, J.; Ze, X.; Yu, X.; Pan, X.; Lin, A.; Zhao, Y.; Zhang, C.; et al. TiO2 nanoparticles induced hippocampal neuroinflammation in mice. PLoS ONE 2014, 9, e92230.
- Disdier, C.; Chalansonnet, M.; Gagnaire, F.; Gaté, L.; Cosnier, F.; Devoy, J.; Saba, W.; Lund, A.K.; Brun, E.; Mabondzo, A. Brain Inflammation, Blood Brain Barrier dysfunction and Neuronal Synaptophysin Decrease after Inhalation Exposure to Titanium Dioxide Nano-aerosol in Aging Rats. Sci. Rep. 2017, 7, 12196.
- Samiei, F.; Shirazi, F.H.; Naserzadeh, P.; Dousti, F.; Seydi, E.; Pourahmad, J. Toxicity of multi-wall carbon nanotubes inhalation on the brain of rats. Environ. Sci. Pollut. Res. Int. 2020, 27, 12096–12111, Erratum in Environ. Sci. Pollut. Res. Int. 2020, 27, 29699.
- Hubbs, A.F.; Porter, D.W.; Mercer, R.R.; Castranova, V.; Sargent, L.M.; Sriram, K. Nanoparticulates. In Haschek and Rousseaux’s Handbook of Toxicologic Pathology, 4th ed.; Haschek, W., Rousseaux, C., Wallig, M., Eds.; Elsevier Press: Amsterdam, The Netherlands, 2023; Volume 3, Chapter 30; ISBN 9780443161537.
- Klein, A.L.; Nugent, G.; Cavendish, J.; Geldenhuys, W.J.; Sriram, K.; Porter, D.; Fladeland, R.; Lockman, P.R.; Sherman, J.H. Nanoparticles as a Tool in Neuro-Oncology Theranostics. Pharmaceutics 2021, 13, 948.
- Thiruvengadam, M.; Rajakumar, G.; Swetha, V.; Ansari, M.A.; Alghamdi, S.; Almehmadi, M.; Halawi, M.; Kungumadevi, L.; Raja, V.; Sabura Sarbudeen, S.; et al. Recent Insights and Multifactorial Applications of Carbon Nanotubes. Micromachines 2021, 12, 1502.
- Yuan, X.; Zhang, X.; Sun, L.; Wei, Y.; Wei, X. Cellular Toxicity and Immunological Effects of Carbon-based Nanomaterials. Part Fibre Toxicol. 2019, 16, 18.
- Donaldson, K.; Aitken, R.; Tran, L.; Stone, V.; Duffin, R.; Forrest, G.; Alexander, A. Carbon nanotubes: A review of their properties in relation to pulmonary toxicology and workplace safety. Toxicol Sci. 2006, 92, 5–22.
- Mercer, R.R.; Scabilloni, J.F.; Hubbs, A.F.; Wang, L.; Battelli, L.A.; McKinney, W.; Castranova, V.; Porter, D.W. Extrapulmonary transport of MWCNT following inhalation exposure. Part Fibre Toxicol. 2013, 10, 38.
- Albini, A.; Pagani, A.; Pulze, L.; Bruno, A.; Principi, E.; Congiu, T.; Gini, E.; Grimaldi, A.; Bassani, B.; De Flora, S.; et al. Environmental impact of multi-wall carbon nanotubes in a novel model of exposure: Systemic distribution, macrophage accumulation, and amyloid deposition. Int. J. Nanomed. 2015, 10, 6133–6145.
- Huang, C.L.; Hsiao, I.L.; Lin, H.C.; Wang, C.F.; Huang, Y.J.; Chuang, C.Y. Silver nanoparticles affect on gene expression of inflammatory and neurodegenerative responses in mouse brain neural cells. Environ. Res. 2015, 136, 253–263.
- Li, X.; Sun, W.; An, L. Nano-CuO impairs spatial cognition associated with inhibiting hippocampal long-term potentiation via affecting glutamatergic neurotransmission in rats. Toxicol. Ind. Health 2018, 34, 409–421.
- Varlamova, E.G.; Turovsky, E.A.; Babenko, V.A.; Plotnikov, E.Y. The Mechanisms Underlying the Protective Action of Selenium Nanoparticles against Ischemia/Reoxygenation Are Mediated by the Activation of the Ca2+ Signaling System of Astrocytes and Reactive Astrogliosis. Int. J. Mol. Sci. 2021, 22, 12825.
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980.
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458.
- Obst, J.; Simon, E.; Mancuso, R.; Gomez-Nicola, D. The Role of Microglia in Prion Diseases: A Paradigm of Functional Diversity. Front. Aging Neurosci. 2017, 9, 207.
- Hristovska, I.; Robert, M.; Combet, K.; Honnorat, J.; Comte, J.C.; Pascual, O. Sleep decreases neuronal activity control of microglial dynamics in mice. Nat. Commun. 2022, 13, 6273.
- Osborn, L.M.; Kamphuis, W.; Wadman, W.J.; Hol, E.M. Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 121–141.
- Cicognola, C.; Janelidze, S.; Hertze, J.; Zetterberg, H.; Blennow, K.; Mattsson-Carlgren, N.; Hansson, O. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimer’s Res. Ther. 2021, 13, 68.
- World Health Organization. Global action plan on the public health response to dementia 2017–2025. Geneva: World Health Organization; 2017. Licence: CC BY-NC-SA 3.0 IGO. Available online: https://apps.who.int/iris/bitstream/handle/10665/259615/9789241513487-eng.pdf?sequence=1 (accessed on 1 December 2022).
- Fong, T.G.; Inouye, S.K. The inter-relationship between delirium and dementia: The importance of delirium prevention. Nat. Rev. Neurol. 2022, 18, 579–596.
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472.
- Dejanovic, B.; Wu, T.; Tsai, M.C.; Graykowski, D.; Gandham, V.D.; Rose, C.M.; Bakalarski, C.E.; Ngu, H.; Wang, Y.; Pandey, S.; et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat. Aging 2022, 2, 837–850.
- Rossi, F.; Bianchini, E. Synergistic induction of nitric oxide by beta-amyloid and cytokines in astrocytes. Biochem. Biophys. Res. Commun. 1996, 225, 474–478.
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol. Dis. 2000, 7, 682–689.
- Combs, C.K.; Karlo, J.C.; Kao, S.C.; Landreth, G.E. Beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188.
- Liao, Y.F.; Wang, B.J.; Cheng, H.T.; Kuo, L.H.; Wolfe, M.S. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 2004, 279, 49523–49532.
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368.
- Park, K.M.; Bowers, W.J. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell. Signal. 2010, 22, 977–983.
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150.
- Goldgaber, D.; Harris, H.W.; Hla, T.; Maciag, T.; Donnelly, R.J.; Jacobsen, J.S.; Vitek, M.P.; Gajdusek, D.C. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7606–7610.
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; Macleod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615.
- Sheng, J.G.; Zhu, S.G.; Jones, R.A.; Griffin, W.S.; Mrak, R.E. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp. Neurol. 2000, 163, 388–391.
- Hoozemans, J.J.; Veerhuis, R.; Janssen, I.; Rozemuller, A.J.; Eikelenboom, P. Interleukin-1beta induced cyclooxygenase 2 expression and prostaglandin E2 secretion by human neuroblastoma cells: Implications for Alzheimer’s disease. Exp. Gerontol. 2001, 36, 559–570.
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611.
- Griffin, W.S.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflamm. 2006, 3, 5.
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360.
- Tachida, Y.; Nakagawa, K.; Saito, T.; Saido, T.C.; Honda, T.; Saito, Y.; Murayama, S.; Endo, T.; Sakaguchi, G.; Kato, A.; et al. Interleukin-1 beta up-regulates TACE to enhance alpha-cleavage of APP in neurons: Resulting decrease in Abeta production. J. Neurochem. 2008, 104, 1387–1393.
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549.
- Ringheim, G.E.; Szczepanik, A.M.; Petko, W.; Burgher, K.L.; Zhu, S.Z.; Chao, C.C. Enhancement of beta-amyloid precursor protein transcription and expression by the soluble interleukin-6 receptor/interleukin-6 complex. Brain Res. Mol. Brain Res. 1998, 55, 35–44.
- Quintanilla, R.A.; Orellana, D.I.; Gonzalez-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257.
- Dugan, L.L.; Ali, S.S.; Shekhtman, G.; Roberts, A.J.; Lucero, J.; Quick, K.L.; Behrens, M.M. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS ONE 2009, 4, e5518.
- Chakrabarty, P.; Ceballos-Diaz, C.; Beccard, A.; Janus, C.; Dickson, D.; Golde, T.E.; Das, P. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J. Immunol. 2010, 184, 5333–5343.
- Spooren, A.; Kolmus, K.; Laureys, G.; Clinckers, R.; De Keyser, J.; Haegeman, G.; Gerlo, S. Interleukin-6, a mental cytokine. Brain Res. Rev. 2011, 67, 157–183.
- Curran, B.; O’Connor, J.J. The pro-inflammatory cytokine interleukin-18 impairs long-term potentiation and NMDA receptor-mediated transmission in the rat hippocampus in vitro. Neuroscience 2001, 108, 83–90.
- Ojala, J.O.; Sutinen, E.M.; Salminen, A.; Pirttila, T. Interleukin-18 increases expression of kinases involved in tau phosphorylation in SH-SY5Y neuroblastoma cells. J. Neuroimmunol. 2008, 205, 86–93.
- Sutinen, E.M.; Pirttilä, T.; Anderson, G.; Salminen, A.; Ojala, J.O. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J. Neuroinflammation 2012, 9, 199.
- Yermakova, A.V.; Rollins, J.; Callahan, L.M.; Rogers, J.; O’Banion, M.K. Cyclooxygenase-1 in human Alzheimer and control brain: Quantitative analysis of expression by microglia and CA3 hippocampal neurons. J. Neuropathol. Exp. Neurol. 1999, 58, 1135–1146.
- Liu, B.; Gao, H.M.; Wang, J.Y.; Jeohn, G.H.; Cooper, C.L.; Hong, J.S. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann. N. Y. Acad. Sci. 2002, 962, 318–331.
- Bishop, A.; Anderson, J.E. No signaling in the CNS: From the physiological to the pathological. Toxicology 2005, 208, 193–205.
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775.
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097.
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065.
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123.e6.
- Brea, D.; Veiga-Fernandes, H. Inflammation in the gut is encoded by neurons in the brain. Nature 2022, 602, 217–218.
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150.
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52.
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? Acc. Chem. Res. 2019, 52, 2026–2035.
- Cheng, Y.; Song, Y.; Chen, H.; Li, Q.; Gao, Y.; Lu, G.; Luo, C. Ferroptosis Mediated by Lipid Reactive Oxygen Species: A Possible Causal Link of Neuroinflammation to Neurological Disorders. Oxidative Med. Cell. Longev. 2021, 5005136.
- Weiss, J.H.; Hartley, D.M.; Koh, J.Y.; Choi, D.W. AMPA receptor activation potentiates zinc neurotoxicity. Neuron 1993, 10, 43–49.
- Narayanan, S.E.; Rehuman, N.A.; Harilal, S.; Vincent, A.; Rajamma, R.G.; Behl, T.; Uddin, M.S.; Ashraf, G.M.; Mathew, B. Molecular mechanism of zinc neurotoxicity in Alzheimer’s disease. Environ. Sci. Pollut. Res. Int. 2020, 27, 43542–43552.
- Ghosh, A.; Comerota, M.M.; Wan, D.; Chen, F.; Propson, N.E.; Hwang, S.H.; Hammock, B.D.; Zheng, H. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2020, 12, eabb1206.
- DeMaagd, G.; Philip, A. Parkinson’s Disease and Its Management: Part 1: Disease Entity, Risk Factors, Pathophysiology, Clinical Presentation, and Diagnosis. Pharm. Ther. 2015, 40, 504–532.
- Mastrangelo, L. The Genetics of Parkinson Disease. Adv. Genet. 2017, 98, 43–62.
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; Chapter 5.
- Wilkaniec, A.; Lenkiewicz, A.M.; Babiec, L.; Murawska, E.; Jęśko, H.M.; Cieślik, M.; Culmsee, C.; Adamczyk, A. Exogenous Alpha-Synuclein Evoked Parkin Downregulation Promotes Mitochondrial Dysfunction in Neuronal Cells. Implications for Parkinson’s Disease Pathology. Front. Aging Neurosci. 2021, 13, 591475.
- Braak, H.; Braak, E. Pathoanatomy of Parkinson’s disease. J. Neurol. 2000, 247 (Suppl. S2), II3–II10.
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211.
- Vaccari, C.; El Dib, R.; Gomaa, H.; Lopes, L.C.; de Camargo, J.L. Paraquat and Parkinson’s disease: A systematic review and meta-analysis of observational studies. J. Toxicol. Environ. Health B Crit. Rev. 2019, 22, 172–202.
- Selmaj, K.W.; Farooq, M.; Norton, W.T.; Raine, C.S.; Brosnan, C.F. Proliferation of astrocytes in vitro in response to cytokines. A primary role for tumor necrosis factor. J. Immunol. 1990, 144, 129–135.
- Merrill, J.E. Effects of interleukin-1 and tumor necrosis factor-alpha on astrocytes, microglia, oligodendrocytes, and glial precursors in vitro. Dev. Neurosci. 1991, 13, 130–137.
- Sedgwick, J.D.; Riminton, D.S.; Cyster, J.G.; Korner, H. Tumor necrosis factor: A master-regulator of leukocyte movement. Immunol. Today 2000, 21, 110–113.
- Leon, L.R. Invited review: Cytokine regulation of fever: Studies using gene knockout mice. J. Appl. Physiol. 2002, 92, 2648–2655.
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFalpha. Science 2002, 295, 2282–2285.
- Pickering, M.; Cumiskey, D.; O’Connor, J.J. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp. Physiol. 2005, 90, 663–670.
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential Regulation of AMPA Receptor and GABA Receptor Trafficking by Tumor Necrosis Factor-. J. Neurosci. 2005, 25, 3219–3228.
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-. Nature 2006, 440, 1054–1059.
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221.
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20.
- Lu, K.T.; Wang, Y.W.; Wo, Y.Y.; Yang, Y.L. Extracellular signal-regulated kinase-mediated IL-1-induced cortical neuron damage during traumatic brain injury. Neurosci. Lett. 2005, 386, 40–45.
- Huang, Y.; Smith, D.E.; Ibanez-Sandoval, O.; Sims, J.E.; Friedman, W.J. Neuron-specific effects of interleukin-1beta are mediated by a novel isoform of the IL-1 receptor accessory protein. J. Neurosci. 2011, 31, 18048–18059.
- Kaur, C.; Sivakumar, V.; Zou, Z.; Ling, E.A. Microglia-derived proinflammatory cytokines tumor necrosis factor-alpha and interleukin-1beta induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct. Funct. 2014, 219, 151–170.
- Vawter, M.P.; Dillon-Carter, O.; Tourtellotte, W.W.; Carvey, P.; Freed, W.J. TGFbeta1 and TGFbeta2 concentrations are elevated in Parkinson’s disease in ventricular cerebrospinal fluid. Exp. Neurol. 1996, 142, 313–322.
- Goris, A.; Williams-Gray, C.H.; Foltynie, T.; Brown, J.; Maranian, M.; Walton, A.; Compston, D.A.; Barker, R.A.; Sawcer, S.J. Investigation of TGFB2 as a candidate gene in multiple sclerosis and Parkinson’s disease. J. Neurol. 2007, 254, 846–848.
- Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. Roles for the TGFbeta superfamily in the development and survival of midbrain dopaminergic neurons. Mol. Neurobiol. 2014, 50, 559–573.
- Tesseur, I.; Nguyen, A.; Chang, B.; Li, L.; Woodling, N.S.; Wyss-Coray, T.; Luo, J. Deficiency in neuronal TGF-beta signaling leads to nigrostriatal degeneration and activation of TGF-beta signaling protects against MPTP neurotoxicity in mice. J. Neurosci. 2017, 37, 4584–4592.
- Ebadi, M.; Sharma, S.K. Peroxynitrite and mitochondrial dysfunction in the pathogenesis of Parkinson’s disease. Antioxid. Redox Signal 2003, 5, 319–335.
- Zhang, L.; Dawson, V.L.; Dawson, T.M. Role of nitric oxide in Parkinson’s disease. Pharmacol. Ther. 2006, 109, 33–41.
- Tsang, A.H.; Chung, K.K. Oxidative and nitrosative stress in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 643–650.
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; Herrero, M.T.; García-Martín, E.; Agúndez, J.A. An Update on the Role of Nitric Oxide in the Neurodegenerative Processes of Parkinson’s Disease. Curr. Med. Chem. 2016, 23, 2666–2679.
- Sircar, E.; Rai, S.R.; Wilson, M.A.; Schlossmacher, M.G.; Sengupta, R. Neurodegeneration: Impact of S-nitrosylated Parkin, DJ-1 and PINK1 on the pathogenesis of Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108869.
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291.
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605.
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: Implications for Parkinson’s disease. FASEB J. 2002, 16, 1474–1476.
- Sriram, K.; Benkovic, S.A.; Hebert, M.A.; Miller, D.B.; O’Callaghan, J.P. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: Key signaling pathway for astrogliosis in vivo? J. Biol. Chem. 2004, 279, 19936–19947.
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: Role of TNF-alpha. FASEB J. 2006, 20, 670–682.
- Sriram, K.; Miller, D.B.; O’Callaghan, J.P. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: Role of tumor necrosis factor-alpha. J. Neurochem. 2006, 96, 706–718.
- Mastroeni, D.; Nolz, J.; Sekar, S.; Delvaux, E.; Serrano, G.; Cuyugan, L.; Liang, W.S.; Beach, T.G.; Rogers, J.; Coleman, P.D. Laser-captured microglia in the Alzheimer’s and Parkinson’s brain reveal unique regional expression profiles and suggest a potential role for hepatitis B in the Alzheimer’s brain. Neurobiol. Aging 2018, 63, 12–21.
- Chou, T.W.; Chang, N.P.; Krishnagiri, M.; Patel, A.P.; Lindman, M.; Angel, J.P.; Kung, P.L.; Atkins, C.; Daniels, B.P. Fibrillar α-synuclein induces neurotoxic astrocyte activation via RIP kinase signaling and NF-κB. Cell Death Dis. 2021, 12, 756.
- Haïk, S.; Marcon, G.; Mallet, A.; Tettamanti, M.; Welaratne, A.; Giaccone, G.; Azimi, S.; Pietrini, V.; Fabreguettes, J.R.; Imperiale, D.; et al. Doxycycline in Creutzfeldt-Jakob disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014, 13, 150–158.
- Watson, N.; Kirby, J.; Kurudzhu, H.; Leitch, M.; MacKenzie, J.; Smith-Bathgate, B.; Smith, C.; Summers, D.; Green, A.J.E.; Pal, S. Impact of the COVID-19 pandemic on Creutzfeldt-Jakob disease surveillance and patient care in the United Kingdom. Eur. J. Neurol. 2022, 29, 1222–1226.
- Aguzzi, A.; Calella, A.M. Prions: Protein aggregation and infectious diseases. Physiol. Rev. 2009, 89, 1105–1152.
- Zerr, I. Laboratory Diagnosis of Creutzfeldt-Jakob Disease. N. Engl. J. Med. 2022, 386, 1345–1350.
- Safar, J.G. Molecular pathogenesis of sporadic prion diseases in man. Prion 2012, 6, 108–115.
- Uttley, L.; Carroll, C.; Wong, R.; Hilton, D.A.; Stevenson, M. Creutzfeldt-Jakob disease: A systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 2020, 20, e2–e10.
- Stoeck, K.; Bodemer, M.; Zerr, I. Pro- and anti-inflammatory cytokines in the CSF of patients with Creutzfeldt-Jakob disease. J Neuroimmunol. 2006, 172, 175–181.
- Gómez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. 2013, 33, 2481–2493.
- Deininger, M.H.; Bekure-Nemariam, K.; Trautmann, K.; Morgalla, M.; Meyermann, R.; Schluesener, H.J. Cyclooxygenase-1 and -2 in brains of patients who died with sporadic Creutzfeldt-Jakob disease. J. Mol. Neurosci. 2003, 20, 25–30.
- Llorens, F.; López-González, I.; Thüne, K.; Carmona, M.; Zafar, S.; Andréoletti, O.; Zerr, I.; Ferrer, I. Subtype and regional-specific neuroinflammation in sporadic creutzfeldt-jakob disease. Front. Aging Neurosci. 2014, 6, 198.
- Guentchev, M.; Siedlak, S.L.; Jarius, C.; Tagliavini, F.; Castellani, R.J.; Perry, G.; Smith, M.A.; Budka, H. Oxidative damage to nucleic acids in human prion disease. Neurobiol. Dis. 2002, 9, 275–281.
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82.
- Szpak, G.M.; Lewandowska, E.; Lechowicz, W.; Wierzba-Bobrowicz, T.; Kulczycki, J.; Bertrand, E.; Pasennik, E.; Dymecki, J. The brain immune response in human prion diseases. Microglial activation and microglial disease. I. Sporadic Creutzfeldt-Jakob disease. Folia Neuropathol. 2006, 44, 202–213.
- McGeer, P.L.; McGeer, E.G. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 459–470.
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; NYGC ALS Consortium; Fagegaltier, D.; Harris, B.T.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177.e5.
- Lall, D.; Lorenzini, I.; Mota, T.A.; Bell, S.; Mahan, T.E.; Ulrich, J.D.; Davtyan, H.; Rexach, J.E.; Muhammad, A.K.M.G.; Shelest, O.; et al. C9orf72 deficiency promotes microglial-mediated synaptic loss in aging and amyloid accumulation. Neuron 2021, 109, 2275–2291.e8.
- Apolloni, S.; Amadio, S.; Parisi, C.; Matteucci, A.; Potenza, R.L.; Armida, M.; Popoli, P.; D’Ambrosi, N.; Volonté, C. Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis. Models Mech. 2014, 7, 1101–1109.
- Maniatis, S.; Äijö, T.; Vickovic, S.; Braine, C.; Kang, K.; Mollbrink, A.; Fagegaltier, D.; Andrusivová, Ž.; Saarenpää, S.; Saiz-Castro, G.; et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 2019, 364, 89–93.
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An (R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609.
- D’Erchia, A.M.; Gallo, A.; Manzari, C.; Raho, S.; Horner, D.S.; Chiara, M.; Valletti, A.; Aiello, I.; Mastropasqua, F.; Ciaccia, L.; et al. Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 2017, 7, 10046.
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340.
- Dols-Icardo, O.; Montal, V.; Sirisi, S.; López-Pernas, G.; Cervera-Carles, L.; Querol-Vilaseca, M.; Muñoz, L.; Belbin, O.; Alcolea, D.; Molina-Porcel, L.; et al. Motor cortex transcriptome reveals microglial key events in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e829.
- Konjevic Sabolek, M.; Held, K.; Beltrán, E.; Niedl, A.G.; Meinl, E.; Hohlfeld, R.; Lassmann, H.; Dornmair, K. Communication of CD8+ T cells with mononuclear phagocytes in multiple sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 1151–1164.
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132 Pt 5, 1175–1189.
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—the plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955.
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269.
- Böttcher, C.; van der Poel, M.; Fernández-Zapata, C.; Schlickeiser, S.; Leman, J.K.H.; Hsiao, C.C.; Mizee, M.R.; Adelia Vincenten, M.C.J.; Kunkel, D.; Huitinga, I.; et al. Single-cell mass cytometry reveals complex myeloid cell composition in active lesions of progressive multiple sclerosis. Acta Neuropathol. Commun. 2020, 8, 136.
- Hu, C.F.; Wu, S.P.; Lin, G.J.; Shieh, C.C.; Hsu, C.S.; Chen, J.W.; Chen, S.H.; Hong, J.S.; Chen, S.J. Microglial Nox2 Plays a Key Role in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2021, 12, 638381.
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748.
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517.
- Fugger, L.; Friese, M.A.; Bell, J.I. From genes to function: The next challenge to understanding multiple sclerosis. Nat. Rev. Immunol. 2009, 9, 408–417.

