An occupational injury describes any injury or illness to a worker as related to their specific work/occupational demands or requirement. It is often a life-altering event that is also considered a form of disability, which is covered under the provisions of the 1990 Americans with Disabilities Act. Occupational injuries and toxicant exposures, particularly to the brain, can lead to the development of neuroinflammation by activating distinct mechanistic signaling cascades that ultimately culminate in the disruption of neuronal function leading to neurological and neurodegenerative disorders. The entry of toxicants into the brain causes the subsequent activation of glial cells, a response known as ‘reactive gliosis.’ Gliosis, a “reactive” state of glia, is a pathological hallmark of all types of central nervous system (CNS) injuries. Advancements in the field of neuroscience, including techniques for high throughput gene expression analysis, morphometric analysis, cytokine and chemokine analysis, advances in neuroimaging and cellular microscopy, have helped identify the possible molecular and neural targets associated with occupational injury or toxicant exposure, bringing the neuroinflammation perspective into place for the implication of gliosis and subsequent progression of neural injury in conditions like TBI and neurodegenerative diseases like AD, PD, CJD, ALS, and MS.
- Alzheimer’s disease
- Amyotrophic lateral sclerosis
- Astrocytes
- Gliosis
- Hydrocarbons
- Neuroinflammation
- Occupational injuries
- Parkinson's disease
- Brain injury
- Traumatic brain injury
- Multiple sclerosis
- Microglia
- Metal toxicity
- Nanoparticles
1. Occupational Brain Injuries
2. Reactive Gliosis in Neural Injury and Neurodegeneration
| Toxicant/Injury/Disease | Brain Area(s) Affected | Reference(s) |
|---|---|---|
| Occupational Toxicants: |
| Toxicant/Injury/Disease | Brain Area(s) Affected | Reference(s) |
|---|---|---|
| Glial Cell Type | Inflammatory Mediator | Cellular (Neural) Effect | Reference(s) | ||||
|---|---|---|---|---|---|---|---|
| M1 Microglia (reactive) Astrocyte (reactive) | TNFA | ↑ Glutaminase ↑ Glutamic acid ↑ NO ↑ Peroxynitrite formation ↑ BACE 1 & 2 (Beta Secretases) ↑ APH1A (Gamma secretase) ↑ Aβ formation ↑ Aβ aggregation ↑ Aβ oligomers ↑ Aβ fibrils/plaques ↑ Aβ toxicity ↑ Tau ↑ P-Tau ↑ P-Tau ↑ P-Tau aggregates ↑ Tau fibrils/NFTs ↑ Synaptic excitotoxicity ↑ Synaptic dysfunction ↑ Neuronal Apoptosis ↓ Aβ clearance ↓ LTP |
[229][230][231][232]][245,246[,247233],248[234],249[,250235,251] | ||||
| 1,1,1-trichloroethane | CTX, CER, HIP | ||||||
| M1 Microglia (reactive) Astrocyte (reactive) | [ | 23][41] | |||||
| IL1B | ↑ APP expression | ↑ Aβ formation ↑ Aβ aggregation ↑ Aβ oligomers ↑ Aβ fibrils/plaques ↑ Aβ toxicity |
Acrylamide | ||||
| 2, 5-Hexanedione | SN | [69][87] | ↑ Tau ↑ P-Tau ↑ P-Tau ↑ P-Tau aggregates ↑ Tau fibrils/NFTs ↑ NO ↑ Peroxynitrite formation ↑ S100B ↑ p38 MAPK ↑ PTGS2 (COX2) ↑ Prostaglandin E2 (PGE2) ↑ Synaptic plasticity ↑ Synaptic dysfunction ↑ Neuronal damage ↑ Neuronal death ↓ LTP |
[229]245[231][236][237][238][239][240][241]244],247,[252[242,253][243,254],255[,256,257,258,259,260] | CER | ||
| M1 Microglia (reactive) Astrocyte (reactive) | [ | 24][42] | |||||
| Acrylamide | SN | [ | IL6 | ↑ APP expression ↑ Tau ↑ P-Tau ↑ P-Tau ↑ P-Tau aggregates ↑ Tau fibrils/NFTs ↑ p38 MAPK ↑ p35 activator ↑ CDK5 |
Chlorpyrifos | STR | [25][43] |
| Cyclohexane | HIP | [26][44] | |||||
| 70 | ] | [ | ↓ LTP |
[245][246][247][248][249][261,262,263,264,265] | |||
| M1 Microglia (reactive) Astrocyte (reactive) |
IL18 | ↑ Aβ formation ↑ Aβ aggregation ↑ Aβ oligomers ↑ Aβ fibrils/plaques ↑ Aβ toxicity ↑ Tau ↑ P-Tau ↑ P-Tau ↑ P-Tau aggregates ↑ Tau fibrils/NFTs ↑ Synaptic dysfunction ↑ Neuronal damage ↑ Neuronal death ↓ LTP |
[250][251][252 | Dichloromethane | FCT, MCT | [27][45] | |
| Fracking Sand Dust | OB, STR, CER | [28][46] | |||||
| 92 | ] | Manganese | GP, SN-R | ||||
| Diesel Exhaust | [ | 29][47] | |||||
| OB, CTX, MB, SN | [ | 75][93] | Manganese | HIP | |||
| Alzheimer’s disease | |||||||
| Alzheimer’s disease | |||||||
| TCT | |||||||
| [ | |||||||
| 53 | |||||||
| ] | |||||||
| [ | |||||||
| 71 | |||||||
| ] | |||||||
| 88 | ] | ||||||
| Diethyldithiocarbamate | ECT, HIP, HYP | [76][94] | |||||
| Iron oxide nanoparticles | OB, HIP, STR | [77][95] | |||||
| Manganese | SN | [78][96] | |||||
| Alzheimer’s disease | |||||||
| Manganese | GP | [79][97] | |||||
| Paraquat + Maneb | LC | [80][98 | FCT, TCT, OCT, CER, BS | [ | HIP | [54][72] | |
| 94 | Alzheimer’s disease | CN, THL, CER, BS | [55][73] | ||||
| Alzheimer’s disease | HIP | [56][74] | |||||
| Alzheimer’s disease | CTX, PUT, AMG | [57][75] | |||||
| Alzheimer’s disease | HIP, FCT, PCT, TCT, CER | [58][76] | |||||
| Alzheimer’s disease | ECT | [59][77] | |||||
| Alzheimer’s-type dementia | TCT | [60][78] | |||||
| Amyotrophic lateral sclerosis | CTX | [ | |||||
| Multiple sclerosis | |||||||
| WM | [ | 65][83] | |||||
| Aluminum | STR, HIP, CTX | [7161] | Parkinson’s disease | SN, LC, DVN | [66][84] | ||
| ] | [ | 89] | |||||
| ] | [ | 266 | ,267,268] | Cypermethrin | STR, SN | [72][90] | |
| Diacetyl | OB | [73 | |||||
| M1 Microglia (reactive) Astrocyte (reactive) |
COX1 | ↑ Inflammation | ][91] | ||||
| [ | Dichlorvos | STR, SN | [74][79] | ||||
| 253 | ] | [ | 269] | ||||
| M1 Microglia (reactive) Astrocyte (reactive) |
NO | ↑ Inflammation ↑ Protein oxidative damage ↑ Lipid oxidation ↑ Tau fibrils/NFTs ↑ Synaptic excitotoxicity ↑ Synaptic dysfunction ↑ Neuronal damage ↑ Neuronal death |
[254][255][256][270,271,272] | [[30][48] | |||
| Oil dispersant (COREXIT EC9500A) | FCT, HIP | [31][49] | |||||
| Silver nanoparticles | FCT, HIP | [32][50] | |||||
| Toluene | HIP | [33][51] | |||||
| Toluene | HIP, CER | [34][52] | |||||
| ] | Toluene | STR | [35][53 | ||||
| Rotenone | ] | ||||||
| STR, SN | [ | 81][99] | Trimethyltin | HIP | [36 | ||
| Rotenone | HIP, CTX | ][54] | |||||
| [ | 82 | ][100] | Trimethyltin | HIP | [37][55] | ||
| Silver nanoparticles | OB | [83][101] | Welding fumes | STR, MB | [38][56] | ||
| Silver nanoparticles | OB | [84][102] | Welding fumes | STR, MB | [39][57] | ||
| Trimethyltin | HIP | [85][103] | Xylene | FCT, CER | [27][45 | ||
| Trimethyltin | ] | ||||||
| HIP | [ | 37][55] | Brain Injuries: | ||||
| Welding fumes | STR, MB | [86][104] | Cerebellar stab injury | CER | [40][58] | ||
| Welding fumes | STR, MB | [87][105] | Cortical stab injury | CTX | [41][59] | ||
| Cortical stab injury | CTX | [42][60] | |||||
| Facial nerve lesion | CTX | [43][61] | |||||
| Forebrain stab lesion | HIP | [44][62] | |||||
| Hippocampal stab wound | Creutzfeldt-Jackob disease | CTX | |||||
| Brain Injuries: | [ | ||||||
| Cortical stab injury | CTX | [42][60] | |||||
| Mild focal brain ischemia | STR | [88][106] | HIP | [45][63] | |||
| Severe focal brain ischemia | CTX | [46][64] | |||||
| Transient global ischemia | HIP | [47][65 |
2.3.2. Gliosis in PD
PD is a chronic and progressive neurodegenerative disease characterized by motor and non-motor dysfunction, with the former being attributable to the loss of nigrostriatal dopaminergic neurons. The clinical motor dysfunction phenotype comprises of a complex of symptoms including muscle rigidity, resting tremor bradykinesia, and postural instability, collectively termed parkinsonism. The non-motor dysfunction typically involves neurons outside of the dopaminergic pathway, and the clinical phenotype is characterized by sleep disruption, cognitive impairment, and depression [268][269][284,285]. Numerous transgenic animal models have been exploited to study PD, including SNCA (PARK1), parkin RBR E3 ubiquitin protein ligase (PRKN/PARK2/Parkin), PTEN-induced kinase 1 (PINK1/PARK6), parkinsonism-associated deglycase (DJ1/PARK7), and leucine-rich repeat kinase 2 (LRRK2/PARK8). These models capture many of the salient features of PD, but none solely recapitulate all the cardinal features of dopaminergic neurodegeneration [270][286]. For example, while exogenous αSYN (SNCA) leads to mitochondrial dysfunction, the overexpression of PRKN rescues mitochondrial dysfunction [271][287]. The degeneration of the nigrostriatal pathway is always associated with extra-nigral dysfunction involving the dorsal motor nucleus of the glossopharyngeal and vagal nerves, thalamic sub-nuclei, amygdala, and the neocortex [258][272][274,288]. The pathology in PD evolves in stages, beginning with lesions of the dorsal IX/X motor nuclei in the medulla oblongata (stage 1), progressing to the gigantocellular reticular nucleus and caudal raphe nuclei in the medulla oblongata and pontine tegmentum (stage 2), substantia nigra pars compacta/SNpc in the midbrain (stage 3), and the cortical areas, mesocortex (stage 4) and neocortex (stages 5 and 6) [273][289]. Further, the non-motor symptoms often precede motor dysfunction by several years or decades [273][289]. Experimental neurotoxic models of PD also include the use of chemical agents, e.g., 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), 6-hydroxydopamine (6-OHDA), as well as pesticides and herbicides, e.g., rotenone and paraquat [274][290]. Multiple lines of investigations have shown that reactive microgliosis is associated with PD (Table 4).| Glial Cell Type | Inflammatory Mediator | Cellular (Neural) Effect | Reference(s) | ||
|---|---|---|---|---|---|
| M1 Microglia (reactive) Astrocyte (reactive) |
TNFA | ↑ SNCA fibrils ↑ SNCA aggregation ↑ Excitatory synaptic transmission ↑ AMPA receptors ↑ Synaptic dysfunction ↑ Neuronal apoptosis ↑ Neuronal degeneration ↑ mEPSC frequency ↑ mEPSC amplitude ↓ LTP ↓ Inhibitory synaptic transmission ↓ GABRA1 receptors ↓ mIPSC amplitude |
[275][276][277][278]282][291,292,293,294[279][280][,295281,296],297[,298] | ||
| M1 Microglia (reactive) Astrocyte (reactive) |
IL1B | ↑ MAPK ↑ SRC Kinase ↑ SNCA fibrils ↑ Apoptosis ↑ Excitotoxicity ↑ Synaptic dysfunction ↑ Neuronal damage ↑ Neuronal death |
[283]299[284][,300285][,301 | ||
| ] | |||||
| 286 | ] | [ | 287][,302,303] | ||
| M1 Microglia (reactive) Astrocyte (reactive) |
TGFB | ↑ Deregulation of ligands ↑ Deregulation of receptors ↑ Neuronal damage ↑ Neuronal death |
[288][289][290][291][304,305,306,307] | ||
| M1 Microglia (reactive) Astrocyte (reactive) |
NO | ↑ Peroxynitrite formation ↑ S-Nitrosylation of proteins ↑ S-Nitrosylation of PARK proteins ↑ Excitotoxicity ↑ Impaired mitochondria ↑ Mitochondrial dysfunction ↑ Neuronal damage ↑ Neuronal death |
[292][293][294][295][296][308,309,310,311,312] | ||
| Transient global ischemia | |||||
| HIP | |||||
| 62 | |||||
| ] | |||||
| [ | |||||
| 80 | |||||
| ] | |||||
| Creutzfeldt-Jackob disease | |||||
| OB, CTX, HIP, AMG | |||||
| [ | |||||
| 63 | |||||
| Transient focal ischemia | SN | [89][107] | |||
| Transient global ischemia | STR, HIP | [90][108] | |||
| Transient MCAO | SN | [91][109 | |||
| [ | 48 | ][66] | |||
| ] | |||||
| ] | |||||
| Traumatic brain injury | CTX | [92][110] | Transient MCAO | STR, HIP, CTX | [49] |
| [ | |||||
| 81 | |||||
| ] | |||||
| Multiple sclerosis | |||||
| Traumatic brain injury | CWM, THL | [93][67] | |||
| CTX | |||||
| [ | |||||
| 64 | |||||
| ] | |||||
| [ | 82 | ] | |||
| [ | 111 | ] | |||
| Traumatic brain injury | CTX | [51][69] | Traumatic brain injury | MCT | [50][68] |
| Traumatic brain injury | HIP | [52][70] | Traumatic brain injury | CTX | [51][69] |
| Neurologic diseases/disorders: | Traumatic brain injury | HIP | [52][70] | ||
| ] | [ | 112] | Neurologic diseases/disorders: | ||
| Alzheimer’s disease | FCT | [95][113] | |||
| Alzheimer’s disease | ITG, MTG | [96][114] | |||
| Alzheimer’s disease | CER | [97][115] | |||
| Alzheimer’s disease | HIP | [98][116] | |||
| Alzheimer’s disease | FCT | [99][117] | |||
| Alzheimer-type dementia | ECT, CCT, TCT | [100][118] | |||
| Amyotrophic lateral sclerosis | CTX, HIP | [101][119] | |||
| Amyotrophic lateral sclerosis | FTCT | [102][120] | |||
| Amyotrophic lateral sclerosis | CC | [103][121] | |||
| Creutzfeldt-Jackob disease | FCT, TCT, STR, TH, CER | [104][122] | |||
| Multiple sclerosis | WM | [65][83] | |||
| Multiple system atrophy | STR | Parkinson’s disease | OB | [67][85] | |
| Schizophrenia with dementia | FCT, TCT | [68][86] |
| Occupational Toxicants: | ||
| [ | ||
| 105 | ||
| ] | ||
| [ | ||
| 123 | ||
| ] | ||
| Parkinson’s disease | ||
| SN | ||
| [ | ||
| 106 | ||
| ] | ||
| [ | ||
| 124 | ||
| ] | ||
| Parkinson’s disease | ||
| SN | ||
| [ | ||
| 107 | ||
| ] | ||
| [ | ||
| 125 | ||
| ] | ||
| Viral Encephalitis | ||
| FCT, TCT, OCT, CER, BS | ||
| [ | ||
| 94 | ||
| ] | ||
| [ | ||
| 112 | ||
| ] | ||
| Wilson’s disease | ||
| FCT, TCT, OCT, CER, BS | [ | 94][112] |
2.1. Gliosis in Brain Injury
TBI is characterized as one of the leading causes of death and disabling occupational injuries [120][121][122][136,137,138], with a high incidence rate in both the military and civilian populations [123][139]. Nearly 5.3 million people live with TBI in the U.S. [120][121][124][125][126][136,137,140,141,142], including work-related TBI. Approximately one in four mild TBI (mTBI) cases in adults are considered work-related [8][28]. However, neuroinflammatory and neurological outcomes of work-related TBI have not been thoroughly investigated. The U.S. Centers for Disease Control and Prevention (CDC) has disseminated the case definitions for TBI using the Ninth and Tenth Revisions of the International Classification of Disease, Clinical Modification (ICD-9-CM and ICD-10-CM codes), commonly used in injury research [124][127][140,143]. Similarly, the Occupational Injury and Illness Classification System were developed by the U.S. Bureau of Labor Statistics to characterize work-related injuries and illnesses. All-encompassing, the terms concussion, mild head injury, mTBI, and cerebral trauma, are used interchangeably to describe the physical damage and the ensuing symptomatic consequences arising from impairment of the brain structure or function [9][128][5,144]. Concussions are usually caused by a blow, bump or jolt to the head. A hit to the body that causes the head and brain to rush back and forth can also cause a concussion [129][145]. Immune cell activation and inflammatory responses are the cardinal features of traumatic injury. However, poor or failed resolution of acute inflammation, and compromise of the defense mechanisms, can contribute to chronic inflammation or an exaggerated systemic inflammatory response syndrome (SIRS) [122][138]. SIRS occurs shortly after trauma or traumatic injury when inflammatory cytokines enlist peripheral leukocytes to the site of the inflamed or injured tissue [122][130][138,146]. Several lines of evidence point to a role for inflammation in the clinical and functional outcomes seen in TBI [9][5]. Inflammatory mediators, e.g., cytokines and chemokines released during traumatic injury can augment the immune response by engaging several immune cells. Both human and animal studies have shown an increase in the expression of high mobility group box 1 protein (HMGB1) following brain injury [9][5], which binds to TLR4 and initiates the inflammatory cascade. The deposition of oxidized phospholipids and oxygenated free-fatty acids has also been shown to cause acute brain injury and induce an inflammatory response [131][147]. In addition to changes in the lipid profile, several other genes are also upregulated following mild and severe TBI. These include antigen-presenting factors (MHC-II, CD86, CD74), cytokines (IL6, IL12, IL10, IL1B, TGFB, IFNG), phagocytosis factors (FCGR, FCGR4, C3, C4), chemotaxis factors (CCL2, CCL4, CXCL1, CXCL4), and astrocytic proteins (GFAP, AQP4) [9][5]. Head injury is a significant risk factor for the initiation and progression of dementia, characterized by the aggregation of beta-amyloid (Aβ) plaques [132][148]. In addition, repeated insults/injuries to the brain heighten the glial response by enhancing microglial or astrocytic cell density and release of inflammatory mediators [133][134][149,150]. Evidence obtained from human post-mortem tissues revealed that repeat injuries generate a more robust glial response, demonstrating that phosphorylated tau and CD68 cell density can be predictors of repeated head injury [135][151]. The primary pathological outcomes associated with TBI have been reported to include focal intracranial hemorrhage, epidural and subdural hematoma, and axonal injury [123][139]. Computed tomography studies also provided evidence of meningeal vascular leakage in approximately 50% of concussed patients who were otherwise considered clinically normal [136][152]. Such vascular changes can potentially lead to secondary lesions that can progressively culminate in neurological or neurodegenerative outcomes. Following TBI, activated microglia/macrophages exhibit different phenotypic features [137][153], commonly referred to as classically activated or M1 (pro-inflammatory) and alternatively activated or M2 (anti-inflammatory) microglia (Figure 1). The inflammatory or anti-inflammatory glial response to TBI is governed by numerous cell-signaling events that are selectively activated depending on the form and severity of the neural insult [108][138][139][126,154,155]. Thus, microglia become rapidly activated in response to CNS injury caused by TBI.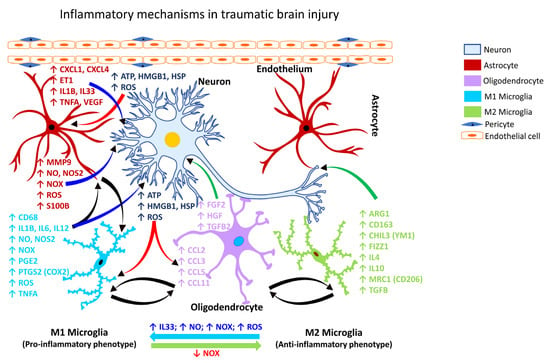
2.2. Gliosis Following Chemical-Induced Neural Injury
2.2.1. Gliosis Associated with Hydrocarbon Exposure
The rapid industrialization and urbanization have brought with it an unprecedented rise in human exposure to an ever-expanding list of hazardous chemicals and pollutants [141][157]. Every year, more than 2000 new chemicals are introduced into the environment, and humans can potentially come in to contact with such chemicals either at the workplace or through daily consumption or use, including via food, cosmetics, pharmaceuticals, cleaning agents, and herbicides/pesticides. In 2008, the Toxicology in the 21st Century (Tox21) Consortium [142][158], a collaboration of federal agencies, including the U.S. Environmental Protection Agency (EPA), the National Institute of Environmental Health Sciences (NIEHS)-National Toxicology Program (NTP), the National Center for Advancing Translational Sciences (NCATS), and the Food and Drug Administration (FDA) came into effect to evaluate and understand the adverse human health risks of commercial chemicals, including pesticides, food additives/contaminants, and medical products [143][144][159,160]. Some of the chemicals of interest to the Tox21 program are polycyclic aromatic hydrocarbons, phthalates, per- and poly-fluoroalkyl substances, bisphenols, and flame retardants [144][160], all of which have potential for adverse human exposures in occupational settings given the consumption demand and high production volumes. Exposure to aliphatic hydrocarbons, particularly n-hexane and halogenated compounds, has been shown to cause a widespread dopaminergic neuronal loss in the substantia nigra and depletion of tyrosine hydroxylase immunoreactivity in the striatum, which was associated with severe gliosis [145][146][161,162]. The effects of several natural and synthetic compounds, including halogenated aromatic hydrocarbons (e.g., biphenyls, dioxins, furans) and polycyclic aromatic hydrocarbons (PAH; e.g., benzo[a]pyrene, benzanthracenes, 3-methylcholanthrene) are mediated by the aryl hydrocarbon receptor (AHR/AhR). The AHR is a ligand-dependent transcription factor that integrates various metabolic cues from the environment, food chain, or microorganisms to regulate transcription in a cell- and ligand-specific manner [147][163]. Increased AHR immunoreactivity is associated with activated microglia in the middle cerebral artery occlusion (MCAO) model of brain ischemia [148][164]. Evaluation of AHR immunoreactivity in human hippocampal post-mortem tissue and its association with reactive astrocytes revealed their greater participation in the development of neurodegenerative diseases, including AD [149][165], suggesting that occupational and environmental exposure to PAH may contribute to the neuropathogenesis seen in many neurodegenerative disorders. Deliberate inhalation of certain volatile hydrocarbons for their mood-altering effects is prevalent among humans. Volatile hydrocarbons are predominantly found in glues, solvents, lighter fluids, gasoline, and paints. High exposure to PAH has been shown to reduce subcortical volume and cause cortical thinning in older people, primarily affecting the parietal, temporal, and insular regions in men, while in women, the frontal and parietal cortical lobes appear to be affected more severely [150][166]. Additionally, PAH formed during the incomplete combustion of organic matter poses a significant risk for firefighters at fire sites [151][167], indicating a potential occupational health risk. Toluene is an aromatic hydrocarbon widely used in various occupational settings, including the paint and adhesive industry; rubber and lumber sector; dry cleaning; automobile and aviation manufacturing; and chemical industries. The highly lipophilic nature of toluene [152][168] can facilitate its entry into the brain after inhalation exposure and target the myelin sheath given it is made up of 70–75% lipids. Thus, the effect of toluene on glial cells, particularly on Schwann cells, oligodendrocytes, and astrocytes are more significant than on the neuronal population, as axonal integrity is relatively well preserved in toluene leukoencephalopathy [152][168]. Toluene also disrupts the differentiation of astrocyte precursors and decreases ATPase activity in astrocytes [153][154][169,170]. Indeed, altered astrocytic function and reactive astrogliosis have been documented in human cases of toluene abuse [155][171]. Toluene is also known to dose-dependently inhibit N-methyl-D-aspartate (NMDA)-mediated excitatory postsynaptic currents (EPSCs) and induce a delayed but persistent reduction in evoked or spontaneous alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)-mediated EPSCs [156][172]. In addition, toluene also inhibits the nicotinic acetylcholine receptor, which has a critical role in brain development [157][173]. In rats inhaling toluene, loss of hippocampal neurons and cerebellar Purkinje cells is reported [158][174], with the latter also causing ‘thinning’ of the cerebellar white matter [158][174]. Chronic or repeated toluene exposure has been shown to induce pro-oxidants, reduce antioxidants, and cause memory impairment [159][175].2.2.2. Gliosis Associated with Metal Exposure
With rapid industrialization, there is an increasing demand for metals. The high-volume production and use of metals are of significant concern for occupational safety due to potential worker exposure and consequent adverse health effects. Chronic workplace exposure to high-production volume metals such as iron (Fe), aluminum (Al), and manganese (Mn) are believed to be associated with an increased risk of neurodegeneration. Fe translocation to the brain is mediated by the transferrin receptor and the solute carrier family 11 member 2 (SLC11A2; also known as divalent metal transporter 1/DMT1 [160][176]. Its prooxidant characteristics elicit ROS generation via the Fenton reaction and Haber-Weiss reaction [161][177], which can subsequently mediate oxidative stress. Free Fe has been known to induce fibrillation and aggregation of alpha-synuclein (αSYN; encoded by the gene SNCA/PARK1) in a dose- and time-dependent manner in cultured cells [162][178]. Emerging evidence also shows that Fe accumulation in the brain accelerates disease progression in AD, but the mechanism through which this occurs is not known [163][179]. Further, the study demonstrated the occurrence of Fe-accumulating microglia that exhibited a dystrophic morphological state. This subset of Fe-accumulating microglia showed augmented expression of the iron storage protein ferritin light chain (FTL) and allograft inflammatory factor 1 (AIF1; also known as ionized calcium-binding adaptor protein 1/IBA1) while downregulating the expression of transmembrane protein 119 (TMEM119) and the purinergic receptor P2Y12 (P2RY12) [163][179]. Al is a potent neurotoxic element involved in the etiology of occupational neurodegenerative disorders. Al causes oxidative stress leading to the deposition of intracellular reactive oxygen species [164][180]. However, the evidence suggesting the role of Al in the development of AD is inconsistent. Chronic oral administration of Al has been shown to increase Aβ levels in the cortex and hippocampus of rats [165][181]. Evidence shows that acute and chronic Al intoxication induces astrogliosis in the motor cortex and the hippocampus [164][180]. Exposure of Tg2576 mice, a model for AD, to Al resulted in increased expression of miRNAs (miR146a and miR125b) demonstrating a proinflammatory response like that seen in the brain of patients with AD [166][167][168][182,183,184]. Mn has been implicated in regulating cellular homeostasis and maintaining physiological functions. While Mn is an essential trace element for normal brain development and function, excess brain Mn is known to be neurotoxic. Occupational exposure to elevated airborne levels of Mn in mining and ferroalloy industries has been reported to cause neurological disorders [169][170][171][172][173][174][175][185,186,187,188,189,190,191], and has been linked to the slowed movement of upper extremities, poor balance and gait coordination, neuropsychological abnormalities, disruption of sleep, cognitive deficits, as well as parkinsonism [176][177][178][192,193,194]. Sustained exposure to low concentrations of Mn has been found to cause Mn-induced parkinsonism [179][195]. Microglial activation has been observed in the substantia nigra pars reticulata (SNpr) and substantia nigra pars compacta (SNpc) of cynomolgus macaques chronically exposed to Mn [180][196]. While the exact mechanisms of Mn transport and neurotoxicity are unclear, several studies suggest that Mn influx to the brain is carrier-mediated [181][182][183][184][197,198,199,200]. Other studies suggest that divalent metal transporters and L-type calcium channels may potentially be involved in the influx of Mn ions across the BBB [185][186][201,202]. Additionally, brain accumulation and toxicity of Mn is thought to be influenced by its elemental speciation. The oxidation state of Mn appears to be critical for its solubility, function, transport, retention, and toxicity [187][188][203,204]. Mn aerosol in ferroalloy industries is reported to exist in mixed oxidation states [189][205] including Mn (0), Mn (II), Mn (III) and Mn (IV). Of these, Mn (II) and Mn (III) are thought to be the predominant forms transported to the brain, where they potentially accumulate [190][206]. Welding fumes (WF) are a complex mixture of toxic metals and gases arising due to the burning of welding electrodes during welding. WF aerosols contain Mn, which is implicated in the development of PD-like neurological dysfunction seen among welders. Particles and aerosols inhaled through the nose and air passages can translocate to the brain via retrograde transport across olfactory neurons and accumulate in deeper brain areas [191][192][193][194][207,208,209,210]. WF may likely follow the same portals for entry into the brain. Alternatively, following deposition in the pulmonary targets, WF particles or soluble metal components of the WF may enter the systemic circulation and be transported to the brain following permeation through the BBB or the circumventricular organs (pituitary gland, median eminence, area postrema, choroid plexus), areas of the brain that are devoid of BBB. Indeed, there is evidence linking Mn and WF exposure to manganism and PD-like manifestations, including neuropsychological and neuropsychiatric disturbances [176][177][195][196][197][198][199][200][201][202][203][192,193,211,212,213,214,215,216,217,218,219]. Further, experimental studies have also shown that WF causes dopaminergic neurotoxicity [39][86][204][205][57,104,220,221].2.2.3. Gliosis Associated with Nanoparticles/Nanomaterials
In recent years, accumulating experimental evidence suggests the potential role of nanoparticles (NPs) in neuroinflammation and brain injury [206][207][208][209][222,223,224,225]. The toxicological effects of nanoparticles depend on their physicochemical properties such as size, shape, and surface charge. The underlying mechanisms of their toxicity are not fully realized, given this is an emerging area of research. Nonetheless, it is thought that much of their effects may be related to their physical interaction with cellular membranes, likely causing membrane disruption, eliciting inflammation, and generating free radicals, including reactive oxygen species (ROS) [210][226].Carbon-Based Nanomaterials
Carbon nanotubes (CNTs) have the structure of tubes made of carbon-based nanomaterials. Carbon nanotubes find application in manufacturing nanocomposites and semiconductors because of their remarkable physicochemical properties, such as high flexibility, good thermal conductivity, low density, and high chemical stability [211][227]. Carbon nanomaterials exhibit a wide variety of toxic effects including inflammatory effects on dendritic cells, ROS generation, DNA damage, pulmonary macrophage activation and inflammation, lysosomal damage, mitochondrial dysfunction, and apoptosis or necrosis-mediated cell death [212][228]. Engineered carbon nanomaterials such as single-walled carbon nanotubes, double-walled carbon nanotubes, and multi-walled carbon nanotubes (MWCNT) have the potential to elicit neurotoxicity due to their small size, ability to aerosolize, and bio-persistence [213][229]. Inhaled MWCNT is distributed to various organs, including the brain [214][230]. Research has shown that MWCNT exhibits spatial association with Aβ fibrils in the brains of mice [215][231]. Further, long-term inhalation of MWCNT has been shown to cause neurological effects, including ROS production, lipid peroxidation, cytochrome c release, and mitochondrial swelling [208][224].Metal Oxide Nanoparticles
Metal oxide nanoparticles (NPs), e.g., copper oxide NP (nano-CuO), iron oxide NP (nano-FeO), silica dioxide NP (nano-SiO2), titanium dioxide NP (nano-TiO2), silver (nano-Ag), selenium NP (nano-Se), and zinc oxide NP (nano-ZnO) are high production volume metal NPs that find application in a variety of industrial processes, manufacturing, agriculture, and nanomedicine. However, due to their small size, large surface area, and potential prooxidant capabilities, there are concerns regarding their toxicologic potential. Exposure of mice to nano-TiO2 has been shown to cause oxidative stress, gliosis, alteration in the expression of genes associated with memory and cognition, as well as the death of hippocampal neurons [206][222]. Inhalation of nano-TiO2 has been shown to augment BBB permeability and inflammatory cytokine production in the brain of aged rats [207][223]. Nano-Ag has been shown to upregulate the expression of inflammatory and antioxidant genes such as interleukin 1 (IL1), C-X-C motif chemokine 13 (CXCL13), macrophage receptor with collagenous structure (MARCO), and glutathione synthetase (GSS) [168][216][184,232]. Exposure to nano-CuO has been shown to decrease the spontaneous excitatory postsynaptic currents (sEPSCs) and miniature EPSCs (mEPSCs) and diminish pre-synaptic and post-synaptic glutamate neurotransmission, indicating reduced long-term potentiation (LTP) and cognitive dysfunction [217][233]. Exposure to nano-Se has been shown to cause enhanced Ca2+ signaling selectively within the astrocytes, increase lactate release, suppress hyperexcitation of neural networks, and activate A2-type astrocytes [218][234].2.3. Gliosis in Neurological Disease States
Microglia play a critical role in regulating CNS physiology in the healthy brain. They are highly ramified structures that maintain direct contact with the dendritic spines, axons, and synapses of neurons, suggesting they are key participants in activity-dependent processes such as the regulation of the synaptic structure and function [219][220][221][222][235,236,237,238]. Like microglia, astrocytes also regulate brain signaling by modulating synapses, regulating homeostasis, transporting nutrients, and maintaining structural support [223][224][239,240].2.3.1. Gliosis in AD
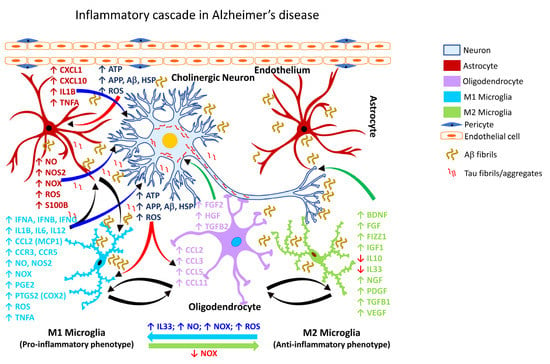
It is estimated that by the year 2050, more than 150 million people worldwide will be impacted by some form of dementia, with AD accounting for nearly 70% of such dementia cases [224][225][240,241]. Chronic neuroinflammation is a prominent feature of AD pathology [226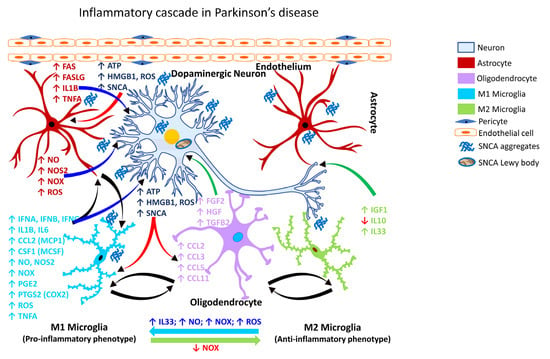
2.3.3. Gliosis in CJD
Creutzfeldt-Jackob disease (CJD) is characterized as a rapidly progressive, fatal, transmissible neurodegenerative disease linked to the accumulation of untreatable prion protein (PrPc) resulting in encephalopathy and neurodegenerative disorders in the CNS [305][306][321,322]. Early diagnosis of CJD remains a major clinical challenge because the manifestation of prion disease at onset are inconsistent and often nonspecific. CJD affects humans and many other mammalian species [221][307][237,323]. Broadly, CJD is categorized into three subtypes: sporadic, inherited, and acquired. The most interesting characteristics of sporadic CJD are pathological features implicated during the development of spongiform change in the cerebral gray matter, which is further associated with the deposition of abnormal forms of prion protein (PrPsc), resulting in cognitive and motor dysfunction [308][324]. While the etiology of sporadic CJD remains unknown, it is hypothesized that a somatic mutation in the prion protein (PRNP) or misfolding of PrPc into PrPsc [309][325] might underlie the etiopathogenesis of CJD. Rapidly progressive dementia is typical in CJD, and the patients generally die within one year after clinical onset [310][326]. Aside from the prion protein-related hypothesis, it is suggested that the cytokine profile in CJD can be pro-inflammatory and anti-inflammatory, with increases seen in the inflammatory cytokine interleukin 8 (IL8) and a decrease in transforming growth factor beta 2 (TGFB2) but no changes in the levels of interleukin 1 beta (IL1B), interleukin 12 (IL12), or tumor necrosis factor alpha (TNFA) [311][327]. The regulatory protein PU1, interleukin 34 (IL34), and CCAAT enhancer binding protein alpha (CEBPA) is also involved in microglial proliferation in CJD [221][312][237,328]. Additionally, prostaglandin-endoperoxide synthase 1 (PTGS1/COX1), prostaglandin-endoperoxide synthase 2 (PTGS2/COX2), and prostaglandin E2 (PGE2) have been reported to be elevated in CJD [313][314][329,330], supporting the involvement of inflammation. Further, it has been shown that the increased inflammatory response seen in CJD is associated with the activation of NFκB and STAT3 signaling pathways [314][330]. Numerous studies have also reported signs of oxidative stress [118][221][315][316][134,237,331,332], and activated microglia in the brains of patients with CJD [317][333].2.3.4. Gliosis in ALS
Amyotrophic lateral sclerosis (also known as Lou Gehrig’s disease) is a progressive neurodegenerative disorder affecting the motor neurons in the cerebral cortex, brainstem, and spinal cord. While the underlying mechanism of the neuronal degeneration remains elusive, a pathological basis involving the ubiquitin-immunoreactive cytoplasmic inclusions has been suggested, which is associated with a robust inflammatory reaction [318][334]. In ALS, mutations in the TAR DNA binding protein (TARDBP) are usually rare despite ALS patients exhibiting cytoplasmic aggregates of TDP43 in the affected brain areas [319][335]. Recent studies have shown that a deficiency of the guanine nucleotide exchange factor C9orf72 (C9orf72) alters the homeostatic gene signature of microglia [320][336] and contributes to the loss of synaptic processes [320][336]. Another line of investigation demonstrated that pharmacological inhibition of the purinergic receptor P2X 7 (P2RX7) decreases microgliosis, inhibits the expression of NF-κB, and attenuates motor neuron death [258][321][274,337]. Further, studies of the spatiotemporal dynamics of microglial activation in SOD1G93A mice have shown that microglial dysfunction precedes the onset of the disease [322][338]. Cumulative evidence over the past two decades shows microglial activation is associated with the degeneration seen in patients with ALS, as assessed by PET analysis. One of the earliest studies revealed diffuse microglial activation in both the motor and non-motor regions of the cerebral cortex [258][319][323][324][325][326][274,335,339,340,341,342]. Additionally, activation of glia in ALS (Figure 4) is associated with marked elevation of ROS, and inflammatory mediators, e.g., PTGS2 (COX2), IL1B, interleukin 6 (IL6), and TNFA [318][334].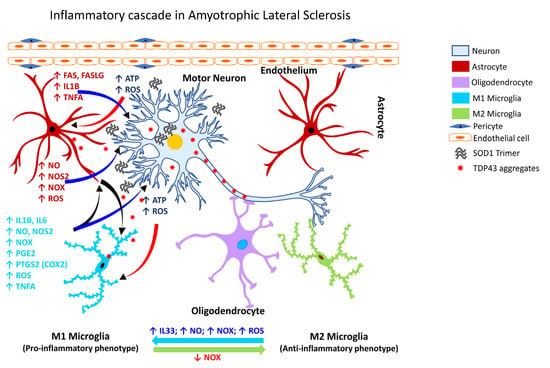
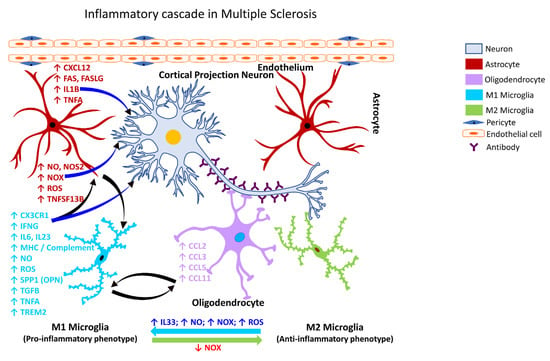
2.3.5. Gliosis in MS
MS is a heterogeneous autoimmune, and complex inflammatory disease of the CNS characterized by demyelinating lesions. The inflammatory outcome in MS is attributed to microglia, however, the extent of their involvement in the disease process and the underlying molecular mechanisms through the mediation of neural injury or damage is still unclear [327][343]. Despite this knowledge gap, the presence of activated microglia at the sites of MS lesions seems to be a common feature. Patients with a progressive course of MS exhibit axonal degeneration and chronic active lesions with microglia typically present at the rim of the pathological lesions [258][328][274,344] in association with complement factors, antibodies, and immune cells [329][330][345,346]. Phagocytic microglia have also been observed in the white matter of the brain tissues obtained from patients with secondary progressive MS [331][347]. Following activation of NADPH Oxidase 2 (CYBB/NOX2), microglia increase their chemotactic signaling and recruit peripheral immune cells to the brain (Figure 5), which play a role in demyelination and axonal damage [332][348]. The intersection of MS pathogenesis and autoreactive T and B lymphocytes has been gaining interest in recent years as they appear to play a role as amplifiers and effectors in MS. Specifically, a subset of T-helper cells, the Th17 cells, appear to play a role in the pathogenesis of MS [333][349] through the secretion of IL12 and related members, interleukin 21 (IL21), and interleukin 23 (IL23). Further, IL21 is known to induce the activation of Th17 cells in an autocrine manner [334][350]. Other factors involved in the inflammatory pathogenesis of MS include polymorphisms in the T-cell receptor beta locus (TRB), cytotoxic T-lymphocyte associated protein 4 (CTLA4), CD6 molecule (CD6), interleukin 10 (IL10), interleukin 2 receptor subunit beta (IL2R), interleukin 4 receptor (IL4R), interleukin 7 receptor (IL7R), C-C motif chemokine receptor 5 (CCR5), interferon-gamma (IFNG), interferon regulatory factor 8 (IRF8), intercellular adhesion molecule 1 (ICAM1), TNFA, TNF receptor superfamily member 1A (TNFRSF1A/TNFRI, vitamin D receptor (VDR), and estrogen receptors 1 and 2 (ESR1 and ESR2) [335][351].