Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Saha Piu | -- | 4986 | 2023-02-06 06:37:36 | | | |
| 2 | Catherine Yang | Meta information modification | 4986 | 2023-02-06 07:28:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Campbell, C.; Kandalgaonkar, M.R.; Golonka, R.M.; Yeoh, B.S.; Vijay-Kumar, M.; Saha, P. Dysregulation of Microbiome–Immunity Interaction in Various Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/40853 (accessed on 07 August 2026).
Campbell C, Kandalgaonkar MR, Golonka RM, Yeoh BS, Vijay-Kumar M, Saha P. Dysregulation of Microbiome–Immunity Interaction in Various Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/40853. Accessed August 07, 2026.
Campbell, Connor, Mrunmayee R. Kandalgaonkar, Rachel M. Golonka, Beng San Yeoh, Matam Vijay-Kumar, Piu Saha. "Dysregulation of Microbiome–Immunity Interaction in Various Diseases" Encyclopedia, https://encyclopedia.pub/entry/40853 (accessed August 07, 2026).
Campbell, C., Kandalgaonkar, M.R., Golonka, R.M., Yeoh, B.S., Vijay-Kumar, M., & Saha, P. (2023, February 06). Dysregulation of Microbiome–Immunity Interaction in Various Diseases. In Encyclopedia. https://encyclopedia.pub/entry/40853
Campbell, Connor, et al. "Dysregulation of Microbiome–Immunity Interaction in Various Diseases." Encyclopedia. Web. 06 February, 2023.
Copy Citation
Gut microbes and their metabolites are actively involved in the development and regulation of host immunity, which can influence disease susceptibility. The role of the microbiome as a protective force is supported by research indicating that immature microbiomes of neonates are more susceptible to invasion by pathobionts. During early years of development, exposure to various microbes shapes the immune system for a lifetime. The gut microbiome has a wide range of metabolic activities, including metabolizing lipids, carbohydrates, and proteins, and participates in maintaining host homeostasis. Therefore, disruption of gut microbiome can lead to conditions as severe as cancer.
gut microbiota dysbiosis
innate immune system
adaptive immune system
1. Gut Microbiota Dysbiosis and Immune Dysregulation
Gut epithelial cells and the mucosa serve as physical barriers against infection and endotoxemia. Gut microbiota metabolites, such as SCFA (short chain fatty acids) and secondary bile acids, also regulate gut permeability via immunomodulation. Of note, another gut-microbiota-derived metabolite inosine, produced by Bifidobacterium and A. muciniphila, heightens Th1 differentiation and effector function of naïve T cells [1]. Gut microbiota-mediated immune responses are essential for preventing intestinal permeability. It is hypothesized that gut microbiota dysbiosis increases intestinal permeability from a ‘leaky gut,’ which allows opportunistic pathogens and their microbial products/toxins to invade the bloodstream and ultimately mount an inflammatory response [2][3][4]. Support for this idea comes from a number of known metabolites, such as phenolic and sulfur-containing compounds, that can harm the intestinal epithelia [5], disrupt intercellular tight junctions [6], and promote bacterial translocation [7]. These consequences, which also include immune cell dysfunction and inability to eliminate the invading pathogens, lead to inflammatory diseases [8][9]. This section of the entry will discuss the microbiota-immune axis in prevalent intra- and extraintestinal diseases (Figure 1 and Table 1).
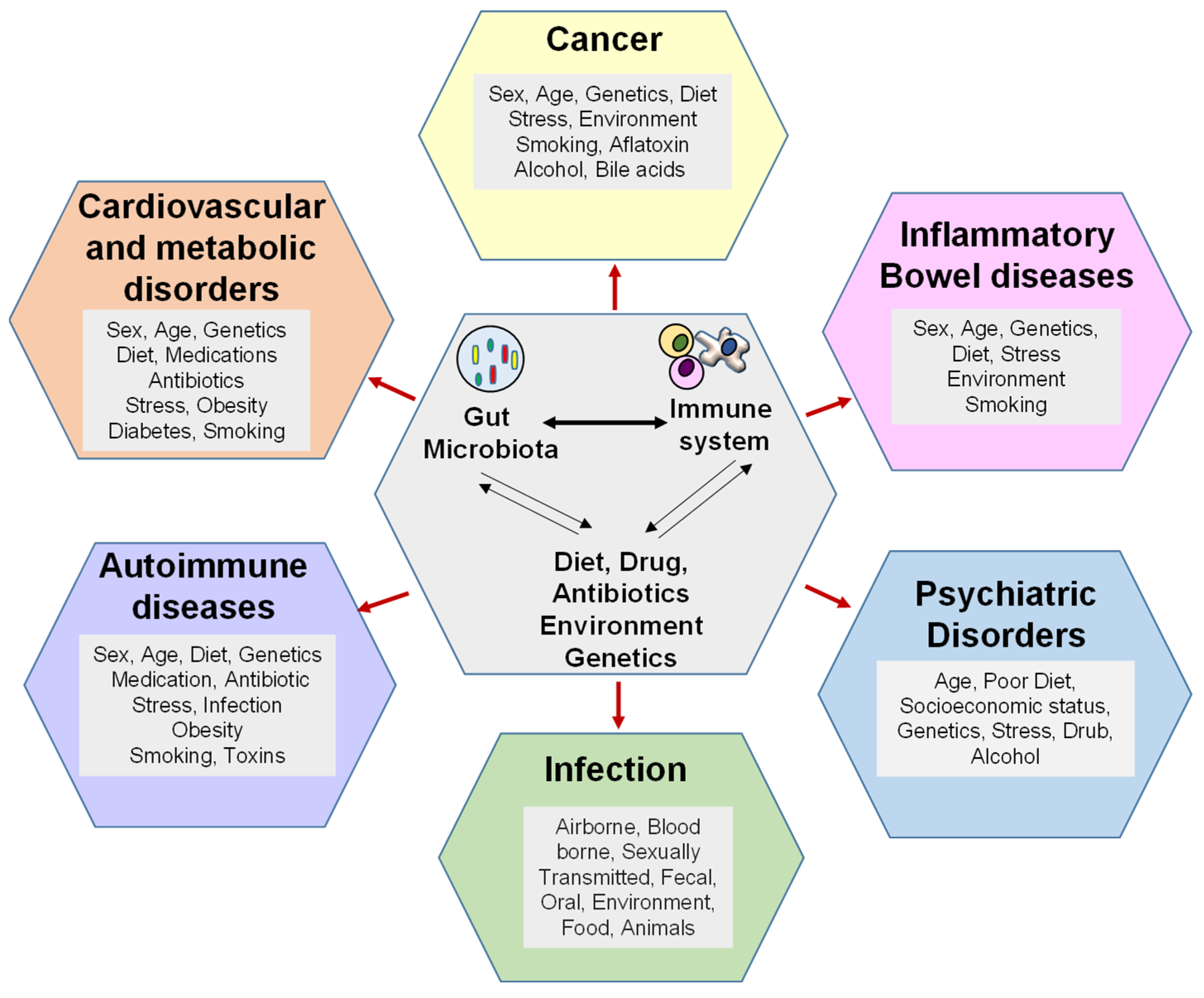
Figure 1. Gut microbiota dysbiosis gives rise to several pathophysiological conditions. Gut microbiota dysbiosis can be induced by diet, antibiotics, and genetic factors. Gut microbiota dysbiosis can cause and sustain cancers, such as colorectal cancer and hepatocellular carcinoma, along with inflammatory diseases, autoimmune conditions, and cardiometabolic disorders. Gut microbiota dysbiosis-induced immune dysregulation is another etiological factor for disease among the many others listed, including age, sex, and medication.
Table 1. Summary of gut microbiota–immune axis in various diseases.
| Diseases | Reference | Findings |
|---|---|---|
| Gastrointestinal Infections | Singer et al., 2019 [10] | Provide resistance against colonization and invasion by pathobiont. |
| Tovaglieri et al., 2019 [11] | Human gut microbiome metabolites induce expression of flagellin (a bacterial protein) increases EHEC motility and epithelial injury. | |
| IBD | Lee and chang, 2021 [12] | Gut microbiota dysbiosis of IBD patients is consistently marked by an overgrowth in Proteobacteria. |
| Furusawa et al., 2013 [13] | SCFA confers protection against IBD by maintaining gut barrier integrity, promoting Treg cell differentiation, and inhibiting histone deacetylases. | |
| Colorectal carcinoma |
Sepich-Poore et al., 2021 [14] | Generation of genotoxin such as Bacteroides fragilis toxin (Bft), cytolethal distending toxin (CDT), and colibactin. |
| Hale et al., 2017 [15] | Bacterial-derived secondary bile acids and hydrogen sulfide promote proinflammatory milieu that increases CRC risk. | |
| Yeoh et al., 2020 [16] | Bacteria such as F. nucleatum can adhere to colon tumors and aggravate tumorigenesis. | |
| Hepatocellular carcinoma | Lin et al., 1995 [17] | Systemic translocation of LPS promotes chronic liver injury and predisposes to HCC. |
| Singh et al., 2018 [18] | Excess butyrate production promotes HCC progression. | |
| Yoshimoto et al., 2013 [19] | Secondary bile acids promote carcinogenesis and impede anti-tumor immunosurveillance in the liver. | |
| Cardiometabolic disease | Cani et al., 2007 [20] Guasch-Ferré et al., 2017 [21] Millard et al., 2018 [22] |
LPS and other microbial ligands drive low-grade chronic inflammation and predispose to CVD. |
| Bacterial trimethylamine and its conversion to trimethylamine-N-oxide in the liver increases the risk of coronary artery disease, metabolic syndrome, stroke, and vascular inflammation. | ||
| Rheumatoid Arthritis |
Scher et al., 2013 [23] | Prevotella spp. Abundance is positively associated with new-onset rheumatoid arthritis. |
| Allergic Diseases |
Fazlollahi et al., 2018 [24] Bunyavanich et al., 2016 [25] |
Gut microbiota dysbiosis increases risk for allergic disease, e.g., food allergy and asthma. |
2. Gastrointestinal Infections
Depending on the context, the gut microbiota can either protect the host or increase risk of infection from exogenous pathogens. The role of the microbiome as a protective force is supported by research indicating that immature microbiomes of neonates are more susceptible to invasion by pathobionts [10]. There are several different mechanisms in which commensals can prevent colonization by pathogens and protect against infections, including competing for resources, releasing bacteriophages, and producing antimicrobial metabolites [26][27][28][29][30]. In contrast, microbiome metabolites, such as 4-methyl benzoic acid, 3,4-dimethylbenzoic acid, hexanoic acid, and heptanoic acid, have been shown to increase colonic epithelial damage, as seen by enterohemorrhagic E. coli in an organ-on-a-chip model [11]. Moreover, supernatant taken from commensal Escherichia albertii can also increase virulence of diarrheagenic E. coli species, resulting in a TLR5-mediated increase in IL-8 and an overall increased pro-inflammatory response by host intestinal cells [31].
Presence of certain commensals and changes in microbiome composition are linked to infection susceptibility by organisms such as Clostridium difficile, Salmonella typhimurium, Escherichia coli, vancomycin-resistant Enterococcus spp., and Citrobacter rodentium [27][28][30][32][33][34]. One of the best examples involves CDI, where innate immune cells are stimulated by C. difficile-toxins through the inflammasome and the TLR4, TLR5, and nucleotide-binding oligomerization domain-containing protein 1 (NOD1) signaling pathways [35][36]. Numerous pro-inflammatory cytokines (such as interleukin (IL)-12, IL-1β, IL-18, interferon gamma (IFN-γ), and tumor necrosis factor α (TNFα)) and chemokines (MIP-1a, MIP-2, and IL-8) are subsequently produced, resulting in increased mucosal permeability, mast cell degranulation, epithelial cell death, and neutrophilic infiltration [37]. Importantly, CDI is usually a result of antibiotic-mediated disruption of the gut microbiota [38]. Eradication of beneficial bacteria in the gut by certain antibiotics, particularly clindamycin, enables C. difficile to flourish [39], resulting in colitis and subsequent diarrhea [40][41]. Besides gut microbiota dysbiosis, immune cell populations, such as Th17- and IL-17-expressing cells, can promote recurrent CDI [42]. Comparatively, IL-33-activated ILCs can prevent CDI [43]. As gut microbiota depletion is a main cause for CDI, interventions that restore microbes could be of therapeutic value.
Prebiotics, such as dietary fiber and their fermented byproducts, i.e., SCFA, are possible treatments for CDI. For instance, dietary fibers, such as pectin, were able to restore gut microbiota eubiosis (denoted by increased Lachnospiraceae and decreased Enterobacteriaceae) and alleviate inflammation following C. difficile-induced colitis [44]. The butyrate producing bacterium Clostridium butyricum was similarly found to protect against CDI by increasing neutrophils, Th1, and Th17 cells in the early phase of infection; this was independent of GPR43 and GPR109a signaling [45]. As mentioned in Section 6.2, CDI can be effectively treated by FMT [46]. FMT is further supported in a prior study that showed that a Microbial Ecosystem Therapeutic, consisting of 33 bacterial strains isolated from human stool, could treat antibiotic-resistant C. difficile colitis [47]. Of note, similar observations were seen when the Microbial Ecosystem Therapeutic was applied to Salmonella typhimurium infection [48]. These findings emphasize that appropriate modulation of the gut microbiota and immune responses are imperative for preventing and fighting against infection.
3. Inflammatory Bowel Diseases
Inflammatory bowel diseases (IBD) develop due to defects in various factors, such as environment, gut microbes, immune system, and genetic factors. IBD involves chronic inflammation of the GIT. Crohn’s disease (CD) and ulcerative colitis (UC) are two distinct clinical conditions of IBD based on histopathological features, location of disease in the GIT, and symptoms [49]. In IBD, mucolytic bacteria and pathogenic bacteria degrade the mucosal barrier and increase the invasion of pathogens into deep intestinal tissues [12][50][51][52]. Alterations in the gut microbiota composition have been highly linked to the development and progression of IBD. IBD patients show reduced populations of Firmicutes and an expansion of Proteobacteria, Bacteroidetes, Enterobacteriaceae, and Bilophila [53][54][55]. In addition, many pro-inflammatory bacterial species are coated with IgA, as seen in IBD patients and colitis mouse models [56][57]. Gut microbes appear to play a direct role in IBD development on the basis of the evidence that germ-free mice are protected against colitis [58]. This is reinforced by the discovery that implantation of gut microbes from IBD mice to germ-free mice resulted in IBD for the latter group [58]. Likewise, dams with IBD can essentially transfer an ‘IBD microbiota’ to the offspring, for which the pups have reduced microbial diversity and fewer class-switched memory B cells and Treg cells in the colon [59]. The strong link between microbiota and IBD has moved forward metagenomic approaches to help better identify diagnostic and therapeutic targets [60].
FMT is proposed as a potential treatment, where treated UC patients were found to have an increased abundance of Faecalibaterium that corresponded with less RORγt+ Th17 cells and more Foxp3+ CD4+ Treg cells [61]. Administration of SCFAs is also thought to be a potential therapeutic for IBD patients [62]. Supporting evidence includes butyrate-mediated inhibition of pro-inflammatory neutrophil responses, i.e., NETs in colitic mice [63]. There are conflicting reports as to whether dietary fiber, the precursor for SCFA, could be a beneficial intervention for IBD patients. On one side, a specific multi-fiber mix was found to counteract intestinal inflammation via increasing IL-10 and Treg cells [64]. Opposingly, the research findings indicate a dichotomy in prebiotic fiber reactions for colitic mice, where pectin could alleviate inflammation compared with inulin, which aggravated the disease pathology [65]. Moreover, the researchers' study suggested that butyrate could be a detrimental microbial metabolite by increasing NLRP3 inflammatory signaling [65]. A probiotic cocktail, comparatively, alleviated inflammation by shifting the gut microbiota to an anti-inflammatory profile which included Akkermansia and Bifidobacterium [66]. These findings collectively indicate that more investigation is required to understand prebiotic fibers and SCFAs in IBD before implementing it in the clinics.
In addition to SCFA, secondary bile acids are implicated in IBD. DCA has been well-established to induce intestinal inflammation [67][68]. This could be due, in part, to bile-acid-mediated inhibition of Paneth cell function [69]. Yet, cholecystectomy-associated secondary bile acids, including DCA, ameliorated colitis in mice by inhibiting monocytes/macrophages recruitment [70]. Moreover, UDCA can also lower colitis severity by preventing the loss of Clostridium cluster XIVa and increasing the abundance of A. muciniphila [71]. The varying effects of bile acids could be related to their chemical structure and potential conjugated moieties. For instance, sulphated secondary bile acids may exert more pro-inflammatory effects compared with their unconjugated counterparts, as seen in IBD patients [72]. Certainly, more metabolomic profiling is necessary to understand the bile acid profile in IBD patients and determine the pro- or anti-inflammatory effects for each type of bile acid. In general, it appears that both SCFA and secondary bile acids have anti-inflammatory effects in the intestine.
Several susceptibility genes that increase risk for IBD have been identified in recent years. Current research is focused on the idea that genetic predisposition, dysbiosis, and environmental factors, such as antibiotics, work in concert toward IBD. Nucleotide-binding oligomerization domain-containing protein 2 (NOD2, an immunological intracellular recognition protein) identifies intracellular muramyl dipeptide (MDP), an integral component of bacterial cell walls [73]. Loss of NOD2 function impairs inhibition of TLR2-mediated activation of NF-κB, resulting in an overactive Th1 response and weakened immunological tolerance to microbes [73]. Moreover, several other genes that increase susceptibility to IBD, including autophagy-related 16-like 1 (ATG16L1), caspase recruitment domain-containing protein 9 (Card9), and C-type lectin domain family 7 member A (CLEC7A), dysregulate T cell responses and create gut microbiota dysbiosis, also contributing to IBD [74][75][76]. Future studies should explore whether there are single nucleotide polymorphisms in genes related to microbial metabolite production for IBD patients.
4. Colorectal Carcinoma (CRC)
A growing body of literature suggests a role for microbiota in the development and progression of cancer. In scenarios where the immune system has maladaptive development, gut microbiota dysbiosis becomes a high risk, and the expansion of certain microbes can result in the production of mutagenic toxins [77]. These genotoxins include Bacteroides fragilis toxin (Bft), cytolethal distending toxin (CDT), and colibactin [14]. However, these highlight only a small number of bacterial-related toxins where more research is needed to identify and understand the carcinogenic potential with the full breadth of gut microbes [14].
Adenomatous and serrated polyps are two precancerous lesions that often progress to colorectal cancer (CRC). In patients with adenomas, several species, including Bilophila, Desulfovibrio, Mogibacterium, and the phylum Bacteroidetes, are increased in the feces, while patients with serrated polyps showed increases in the taxa Fusobacteria and class Erysipelotrichia [15]. Fusobacterium nucleatum (F. nucleatum) is characterized as an important microbe in CRC progression [78][79]. F. nucleatum promotes TLR4 signaling and E-cadherin/β-catenin signaling, ultimately leading to activation of NF-κB and reduced miR-1322 expression [80]. Regulatory micro-RNAs, such as miR-1322, can directly regulate the expression of CCL20, a cytokine that promotes CRC metastasis [78]. Other literature points to F. nucleatum adhesin A (FadA) as a key virulence factor that allows F. nucleatum to adhere, invade, and erode the colonic epithelia [16]. More recently, one study found that F. nucleatum can promote CRC by suppressing anti-tumor immunity through activation of the inhibitory receptors CEACAM1 and TIGIT1, which downregulate NK cells and T cells [81]. The F. nucleatum strain Fn7-1 was also demonstrated to aggravate CRC development by elevating Th17 responses [82]. These findings on F. nucleatum are alarming because this is a SCFA-producing bacterium [83], and SCFA have been, in general, highlighted as a potential therapeutic avenue for many inflammatory diseases. F. nucleatum predominantly produce acetate and butyrate, where it was recently suggested that F. nucleatum induces Th17 via free fatty acid receptor 2 (FFAR2), a SCFA receptor [82]. Yet, loss of FFAR2 in mice aggravated tumor bacterial load and over activated DCs, eventually promoting T cell exhaustion [84]. Moreover, butyrate from dietary fiber was found to be less metabolized in CRC cells because of the Warburg effect, allowing it to act as an HDAC inhibitor and promote acetylation of genes related to apoptosis [85]. These findings emphasize that the pathologic effects of F. nucleatum could be SCFA-independent, but further studies are needed to determine this possibility.
Another proposed mechanism in the development of CRC suggests that excessive dietary intake of sugars, proteins, and lipids could promote the growth of bile-tolerant microbes, which increase production of secondary bile acids, such as DCA and LCA, and by-products, such as hydrogen sulfide. Excessive secondary bile acids are genotoxic and may produce a pro-inflammatory environment that could promote the development of CRC [15]. In particular, DCA can stimulate intestinal carcinogenesis by activating epidermal growth factor receptor-dependent release of the metalloprotease ADAM-17 [86]. DCA also activates β-catenin signaling [87] and drives malignant transformations in Lgr5-expressing (Lgr5+) cancer stem cells [88] for CRC growth and invasiveness. However, bacteria associated with secondary bile acid production, i.e., Clostridium cluster XlVa, were significantly decreased in IBD patients, which was accompanied by reduced transformation of primary to secondary bile acids [89]. In addition to bile acids, the gut microbial metabolite folate can worsen CRC pathogenesis by triggering AhR signaling and expanding Th17 levels [90]. Similar to SCFA, more investigation is needed to discern the potential pro-tumorigenic effects of gut-microbiota-derived bile acids.
There are distinct microbiota-dependent immunological responses in CRC. In terms of innate immune responses, A. muciniphila enrichment facilitated M1 macrophage polarization in an NLRP3-dependent manner that suppressed colon tumorigenesis [91]. Likewise, intestinal adherent E. coli can increase IL-10-producing macrophages, which limits intestinal inflammation and restricts tumor formation [92]. In terms of adaptive immunity, microbial dysbiosis hyperstimulates CD8+ T cells to promote chronic inflammation and early T cell exhaustion, which contributes to colon tumor susceptibility [93]. Intestinal cancer cells can also respond to the microbiota by inducing calcineurin-dependent IL-6 secretion, which promotes tumor expression of the co-inhibitory molecules B7H3/B7H4 that diminish anti-tumor CD8+ T cells [94]. Comparatively, introduction of Helicobacter hepaticus induced T follicular helper cells that restored anti-tumor immunity in a mouse CRC model [95]. Compared with macrophages and Th17 cells, γδ T cells and resident memory T cells were found at lower frequencies in the colonic tissue of CRC patients [96]. It would be interesting to investigate whether an immune cell panel could be developed for early diagnosis of CRC.
5. Hepatocellular Carcinoma (HCC)
Hepatocellular carcinoma (HCC), the most common primary liver cancer, is the fourth leading cause of cancer-related mortality worldwide [97]. The main etiology for HCC pathogenesis comes from pre-existing liver diseases, such as nonalcoholic fatty liver disease (NAFLD) and steatohepatitis, that lead to cirrhosis [98]. This is further complicated by other concomitants in NAFLD patients, including insulin resistance, obesity, and metabolic disorders that further promote hepatic inflammation and tumorigenesis through IL-6 and TNF-α [99]. The liver is the ‘first stop’ for venous blood coming from the intestines, making it vulnerable to the gut microbiota via microbial translocation across the intestinal–epithelial barrier or contact with absorbed microbial metabolites [100]. The aforementioned well-known effects of gut microbiota dysbiosis, including disruption of gut barrier, translocation of microbes into the bloodstream, and subsequent inflammatory immune responses via induction of PRRs by PAMPs, such as LPS, are strongly correlated to the pathogenesis of NAFLD, liver cirrhosis, and HCC [17][100]. While it has long been thought that gut microbiota dysbiosis precedes the development of HCC, this causal relationship has not been explored in depth until more recently. Behary, Raposo et al. recently found, before HCC progression, that gut microbiota dysbiosis is in tandem with early onset liver injury that is followed by an LPS-dependent Th1- and Th17-mediated cytokine response [101]. Further investigation should determine whether gut microbiota dysbiosis is a cause or consequence in the liver injury preceding HCC.
Increased Enterobacteriaceae and Streptococcus and reduction in Akkermansia, alongside elevated levels of inflammatory mediators, such as CCL3, CCL4, CCL5, IL-8, and IL-13, have been noted in patients with NAFLD-associated HCC [102]. A more recent study found decreased abundance of SCFA-producing bacteria and increased LPS-producing bacteria in patients with cirrhosis-induced HCC but no significant evidence of gut microbiota dysbiosis in other liver diseases, such as hepatitis C, hepatitis B, or alcoholic liver disease [103]. Broadly speaking, however, it should be noted that altered microbial populations observed among multiple studies are not consistent with each other [102][104][105][106]. Furthermore, while it is generally thought that SCFAs produced by gut microbes have several benefits for humans, it was recently discovered that inulin, a precursor of the SCFA butyrate, can promote the progression to HCC in genetically altered dysbiotic mice [18]. Other studies have focused on the impact of microbial metabolites on HCC. For instance, a high-fat diet led to gut overgrowth of Gram-positive organisms that generate secondary bile acids, i.e., DCA [19]. DCA can work in concert with lipoteichoic acid to activate TLR2 and subsequently downregulate anti-tumor immunity, creating a microenvironment favorable for the development of HCC [107][108]. Overall, it appears that gut microbiota metabolites are potentially pro-tumorigenic for the liver.
6. Cardiovascular Disease
Cardiovascular disease (CVD) is heavily linked to metabolic syndrome, a condition which involves a set of interrelated diseases—mainly atherosclerosis, NAFLD, hypertension, and type II diabetes mellitus (TIIDM)—that arise from chronic, low-grade inflammation [109]. Many cells with high metabolic activity, such as parenchymal cells in the liver and pancreas, adipocytes, and skeletal myocytes, participate in extensive crosstalk with immune cells. Any perturbation of the microbiome has the potential to alter host immune function and, by extension, may have the ability to cause or alter disease processes in metabolically active tissues. The recognition of LPS and other microbial PAMPs by PRRs are thought to be a key driver in this low-grade inflammatory state [20]. Trimethylamine-N-oxide (TMAO), a microbial co-metabolite, is also noted to cause low-grade inflammation through NF-κB signaling, inflammasome activation, and increased production of free radicals [110][111]. Furthermore, TMAO leads to atherosclerosis and, thus, heart disease by impairing cholesterol metabolism in macrophages and contributing to the formation of foam cells [112]. Indeed, higher serum TMAO is correlated with increased risk of atherosclerosis, coronary artery disease, stroke, and vascular inflammation [21][22], and TMAO is currently being considered as a biomarker for adverse cardiovascular events [113]. More recent research has discovered phenylacetylglutamine (PAGln) as a microbial metabolite related to CVD via adrenergic receptor activation and pro-thrombotic effects [114][115]. There are multiple potential emerging roles for PAGln in cardiovascular medicine, such as being used as a diagnostic marker or even as a predictor for responsiveness to β-blocker therapy for CVD patients [115].
7. Diabetes
Diabetes mellitus is a disease separated into two classes: type I diabetes mellitus (TIDM) involves autoimmune destruction of pancreatic islet cells, while type II diabetes mellitus (TIIDM) involves acquired insulin insensitivity. Though much research involving microbiota and diabetes revolves around TIIDM and obesity, it has been shown that increasing dietary SCFA consumption can lead to altered microbiota and distinct immune profiles in TIDM patients [116]. Increasing dietary SCFAs, such as butyrate and acetate, were also shown to work synergistically to confer protection against autoreactive T cell populations and TIDM in mice [117]. Comparatively, administration of Parabacteroides distasonis accelerated the development of T1DM in a mouse model, and this was because of aberrant immune responses, including elevated CD8+ T cells and decreased Foxp3+ CD4+ Treg cells [118]. Of note, dysregulated bile acid metabolism was found to be a potential predisposing factor for islet autoimmunity and type 1 diabetes [119].
The microbiome and immune systems are both heavily involved in the pathogenesis of TIIDM. Branched-chain amino acids are produced by Prevotella copri (P. copri) and Bacteroides vulgatus spp., and P. copri directly induces insulin resistance in mouse models [120][121]. Depletion of commensal A. muciniphila compromises the intestinal barrier, resulting in translocation of endotoxin into the bloodstream and subsequent activation of CCR2+ monocytes. This results in conversion of pancreatic B1a cells into 4BL cells, which release inflammatory mediators and cause reversible or irreversible insulin resistance [122]. On the other hand, microbial metabolites, such as linoleic acid and docosahexaenoic acid, have protective effects against insulin resistance and TIIDM through anti-inflammatory effects and prevention of lipotoxicity [123]. FMT has also been shown to reduce fasting blood glucose levels and decrease insulin resistance in mice with TIIDM [124]. Furthermore, some of the therapeutic effects of several anti-diabetic drugs can be due, in part, to their ability to alter the microbiota [125][126][127].
8. Hypertension
Several studies have observed significantly altered microbiome compositions between normotensive and hypertensive mice, though specific microbial profiles in hypertensive mice are dependent on the hypertension model used [128][129][130][131]. In the angiotensin II model of hypertension, lack of microbiota in germ-free mice protected against hypertension partly by decreasing inflammatory cell populations in the blood [132]. Yet, germ-free mice were more prone to kidney injury following an angiotensin II and high-salt diet combination regimen [133]. Furthermore, reintroduction of microbiota to hypotensive germ-free mice re-established vascular contractility [134]. Generally, the microbiota composition differs between hypertensive and normotensive animals and, interestingly, cross-fostering hypertensive pups with normotensive dams can reduce blood pressure in the former group [135]. Similar to CVD, the gut metabolite TMAO also has relevance to hypertension. A recent study discovered TMAO exacerbated vasoconstriction via ROS in angiotensin II-induced hypertensive mice [136]. In a similar manner, high-salt-induced DC activation is associated with microbial dysbiosis-mediated hypertension [137]. Comparatively, the ketone body β-hydroxybutyrate has been shown to be decreased in high-salt-fed hypertensive rats; rescuing with the β-hydroxybutyrate precursor 1,3 butanediol decreased blood pressure and kidney inflammation through prevention of the NLRP3-mediated inflammasome [138]. While HSD has been shown elsewhere to decrease Lactobacillus spp. and induce Th17 cell populations, this appears to be through a distinctly different mechanism [139].
9. Rheumatoid Arthritis
The pathogenesis of rheumatoid arthritis (RA), a systemic autoimmune disease characterized primarily by inflammation of joints, is becoming more understood. RA is a multifactorial disease with multiple identified alleles and environmental factors conferring increased susceptibility to the disease. A potentially important microbial genus in the development of RA is Prevotella. This was first identified in 2013 by Scher et al., which found that patients with new onset RA had significantly increased abundance of Prevotella spp., particularly Prevotella copri, compared with healthy controls [23]. However, the Prevotella population did not increase in patients with chronic RA [23]. Since then, multiple studies have found further correlations between various Prevotella species and RA [140][141][142]. However, it is unclear whether Prevotella spp. itself contributes to the pathogenesis of RA, or the immunological environment created by RA increases abundance of Prevotella in the gut.
Other notable bacterial shifts in the gut microbiota for RA patients include a bloom in Proteobacteria, Clostridium cluster XlVa, and Ruminococcus, which were correlated with less CD4+ T cells and Treg cells [143]. Using the K/BxN autoimmune arthritis model, it was found that SFB-mediated cytotoxic T lymphocyte antigen-4 (CTLA-4) reduction caused autoreactive T follicular helper cells [144][145]. The accumulation of T follicular helper cells and Th17 cells in arthritis appears to be age-dependent [146], which helps to explain why RA is found mostly in the older population. Interestingly, though, the gut microbiota seems to predominantly affect T follicular helper cells, not Th17 cells, as confirmed by antibiotic treatment of the K/BxN autoimmune arthritis model [147]. Of note, it was recently reported that collagen-induced RA in mice causes an aberrancy in circadian rhythmic patterns in the gut microbiome, resulting in reduced barrier integrity due to an alteration in circulating microbial-derived factors, such as tryptophan metabolites [148].
SCFAs, specifically butyrate, have been proposed as a therapeutic option for RA. Butyrate supplementation was found to promote Treg cells by inhibiting HDAC expression, and it downregulated pro-inflammatory cytokine genes in RA [149]. Moreover, butyrate alleviated arthritis by directly inducing the differentiation of functional follicular Treg cells in vitro by enhancing histone acetylation via HDAC inhibition [150]. Furthermore, butyrate reduced arthritis severity by increasing the levels of AhR ligands, i.e., serotonin-derived metabolite 5-hydroxyindole-3-acetic acid, where AhR activation supported regulatory B cell function [151]. In addition to SCFA, the gut-microbiota-derived metabolites LCA, DCA, isoLCA, and 3-oxoLCA were also very recently found to exhibit anti-arthritis effects. Specifically, isoLCA and 3-oxoLCA inhibited Th17 differentiation and promoted M2 macrophage polarization [152]. These effects of secondary bile acids could be synergized with Parabacteroides distasonis probiotic supplementation [152]. The newfound findings of secondary bile acids are monumental and need additional investigation.
10. Allergic Diseases
Allergies occur when the immune system becomes hypersensitized to nonpathogenic foreign antigens. Common hypersensitivities include allergic rhinitis, food allergy, eczema, atopic dermatitis, and asthma. Several factors responsible for the development of allergies, such as reduced microbial exposure, cesarean delivery, diet, and antibiotic use are strongly linked to changes in gut microbiome composition [153][154][155][156]. Gut microbiota dysbiosis, in turn, increases risk for allergies, particularly food allergies [24][25]. Dysbiosis induced by antibiotic use is sufficient to increase allergic symptoms, elevate intestinal inflammation, and disrupt gut mucosal tight junction in sensitized mice [157]. A high-fat diet generally has effects similar to antibiotics, causing gut microbiota dysbiosis and subsequently increasing risk for food allergies [158]. Changes in gut microbiota composition immediately after birth, when the microbiome is still establishing, appears to have a particularly large impact on the development of allergic diseases later in life [159]. Of note, the vaginal microbiota can also reflect allergy risk, where Lactobacillus-dominated vaginal microbiota clusters were related to infant serum IgE status at 1 year of age [160].
Several studies reinforce the concept that dysbiosis is heavily linked to allergic disease, especially asthma. Individuals with atopic asthma have significantly higher fecal levels of Lactobacillus and E. coli compared with healthy individuals [161]. In terms of microbiota metabolites, 12,13-diHOME (a relatively uncharacterized linoleic acid) is commonly found in neonates at high risk for asthma [162]. It was recently found that the bacterial epoxide hydrolase, which produces 12,13-diHOME, is also higher in concentration during pulmonary inflammation, and 12,13-diHOME reduced Treg cells in the lung [163][164]. Comparatively, the AhR ligand tetrachlorodibenzo-p-dioxin was able to attenuate delayed-type hypersensitivity by inducing Treg cells, suppressing Th17 cells, and reversing gut microbiota dysbiosis [165]. Likewise, individuals with higher fecal SCFAs, such as butyrate and propionate, early in life had markedly decreased risk for development of asthma and atopy [166]. Of potential therapeutic value, SCFA supplementation could modulate T cells and DCs to alleviate asthma [167]. Similarly, maternal supplementation with dietary fiber or acetate was shown to protect neonates from asthma by promoting acetylation of the Foxp3 gene [168]. Dietary fiber feeding also gave protection from food allergens via retinal dehydrogenase activity in CD103+ DCs [169]. Of note, the dietary fiber inulin was recently found to promote allergen- and helminth-induced type 2 inflammation, and this was bile-acid dependent [170]. Overall, it appears that the influence of gut microbiota on allergies is highly regulated by metabolites, but each microbial product has independent effects that can either promote or demote hypersensitivity.
11. Psychiatric Disorders: The Gut–Brain Axis
The aforementioned information describes the gut microbiota to influence both intra- and extraintestinal diseases. One other organ that the gut microbiota can impact is the brain where a ‘stressed gut’ is becoming more recognized as a pathologic entity in several neurological disorders. For pre-term infants with an immature gut microbiota, Klebsiella overgrowth has been found to be highly predictive for brain damage and is associated with a pro-inflammatory immunological tone [171]. Parkinson’s disease is marked by an accumulation of alpha-synuclein in the gut, and patients often suffer from a leaky gut due to microbiota dysbiosis with higher populations of Prevotellaceae [172]. These symptoms can be reversed by administering probiotics [173][174]. Recently, the idea that microbiota shapes mental health has started gaining traction. Taxonomic and metabolic signatures have been proposed as a biomarker for stratifying major depressive disorder into mild, moderate, and severe symptom categories [175]. Several studies studying differences in microbiota between those who are mentally healthy and those with mental health disorders, such as anxiety and/or depression, have suggested that microbial colonization before and after birth plays a major role later in life. For instance, maternal stress can induce abnormal neurodevelopment in the offspring, which has been marked with a significant reduction of Bifidobacterium spp. [176]. Moreover, neonates delivered by C-section, as opposed to vaginal birth, have a greater risk of developing psychosis later in life [173][177]. Impressively, early-life oxytocin treatment can minimize behavior deficits seen in C-section delivered pups [178].
A cocktail of broad-spectrum, gut-microbiota-depleting antibiotics, specifically at the postnatal and weaning stages, can cause long-lasting effects of anxiety-related behavioral outcomes into adolescence and adulthood [179]. A recent elegant study by Li et al. delineated that infant exposure to antibiotics resulted in anxiety- and depression-like behaviors and memory impairments that were concurrent with an increase inflammatory milieu; similar findings were seen following long-term antibiotic treatment at the adolescent and adult stages in mice [180]. Early-life disruption of the gut microbiota could also cause sex-specific anxiety-like behavior, where LPS treatment to Wistar rats resulted in less social interaction in males compared with the females, who had an increase in social behavior [181]. It is noteworthy that FMT from an ‘aged microbiome’ to germ-free mice decreased SCFAs, and this was associated with cognitive decline [182]. The gut microbiota–immunity–brain axis is still in its nascency and requires investigation to establish mechanisms involved in immune regulation responsible for behavioral abnormalities and neurological disorders. However, it must be emphasized to look at other microorganisms besides bacteria because mucosal fungi were found to promote social behavior through complementary Th17 immune mechanisms [183].
References
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489.
- Ahmadi, S.; Razazan, A.; Nagpal, R.; Jain, S.; Wang, B.; Mishra, S.P.; Wang, S.; Justice, J.; Ding, J.; McClain, D.A.; et al. Metformin Reduces Aging-Related Leaky Gut and Improves Cognitive Function by Beneficially Modulating Gut Microbiome/Goblet Cell/Mucin Axis. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, e9–e21.
- Stevens, B.R.; Goel, R.; Seungbum, K.; Richards, E.M.; Holbert, R.C.; Pepine, C.J.; Raizada, M.K. Increased human intestinal barrier permeability plasma biomarkers zonulin and FABP2 correlated with plasma LPS and altered gut microbiome in anxiety or depression. Gut 2018, 67, 1555–1557.
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.C.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P.; et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 2017, 21, 455–466.
- Hamer, H.M.; De Preter, V.; Windey, K.; Verbeke, K. Functional analysis of colonic bacterial metabolism: Relevant to health? Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1–G9.
- McCall, I.C.; Betanzos, A.; Weber, D.A.; Nava, P.; Miller, G.W.; Parkos, C.A. Effects of phenol on barrier function of a human intestinal epithelial cell line correlate with altered tight junction protein localization. Toxicol. Appl. Pharmacol. 2009, 241, 61–70.
- Cox, L.M.; Yamanishi, S.; Sohn, J.; Alekseyenko, A.V.; Leung, J.M.; Cho, I.; Kim, S.G.; Li, H.; Gao, Z.; Mahana, D.; et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 2014, 158, 705–721.
- Jacobs, M.C.; Haak, B.W.; Hugenholtz, F.; Wiersinga, W.J. Gut microbiota and host defense in critical illness. Curr. Opin. Crit. Care 2017, 23, 257–263.
- Van der Poll, T.; van de Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 2017, 17, 407–420.
- Singer, J.R.; Blosser, E.G.; Zindl, C.L.; Silberger, D.J.; Conlan, S.; Laufer, V.A.; DiToro, D.; Deming, C.; Kumar, R.; Morrow, C.D.; et al. Preventing dysbiosis of the neonatal mouse intestinal microbiome protects against late-onset sepsis. Nat. Med. 2019, 25, 1772–1782.
- Tovaglieri, A.; Sontheimer-Phelps, A.; Geirnaert, A.; Prantil-Baun, R.; Camacho, D.M.; Chou, D.B.; Jalili-Firoozinezhad, S.; de Wouters, T.; Kasendra, M.; Super, M.; et al. Species-specific enhancement of enterohemorrhagic E. coli pathogenesis mediated by microbiome metabolites. Microbiome 2019, 7, 43.
- Lee, M.; Chang, E.B. Inflammatory Bowel Diseases (IBD) and the Microbiome-Searching the Crime Scene for Clues. Gastroenterology 2021, 160, 524–537.
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450.
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science 2021, 371, eabc4552.
- Hale, V.L.; Chen, J.; Johnson, S.; Harrington, S.C.; Yab, T.C.; Smyrk, T.C.; Nelson, H.; Boardman, L.A.; Druliner, B.R.; Levin, T.R.; et al. Shifts in the Fecal Microbiota Associated with Adenomatous Polyps. Cancer Epidemiol. Biomark. Prev. 2017, 26, 85–94.
- Yeoh, Y.K.; Chen, Z.; Wong, M.C.S.; Hui, M.; Yu, J.; Ng, S.C.; Sung, J.J.Y.; Chan, F.K.L.; Chan, P.K.S. Southern Chinese populations harbour non-nucleatum Fusobacteria possessing homologues of the colorectal cancer-associated FadA virulence factor. Gut 2020, 69, 1998–2007.
- Lin, R.S.; Lee, F.Y.; Lee, S.D.; Tsai, Y.T.; Lin, H.C.; Lu, R.H.; Hsu, W.C.; Huang, C.C.; Wang, S.S.; Lo, K.J. Endotoxemia in patients with chronic liver diseases: Relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J. Hepatol 1995, 22, 165–172.
- Singh, V.; Yeoh, B.S.; Chassaing, B.; Xiao, X.; Saha, P.; Aguilera Olvera, R.; Lapek, J.D., Jr.; Zhang, L.; Wang, W.B.; Hao, S.; et al. Dysregulated Microbial Fermentation of Soluble Fiber Induces Cholestatic Liver Cancer. Cell 2018, 175, 679–694.
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101.
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772.
- Guasch-Ferre, M.; Hu, F.B.; Ruiz-Canela, M.; Bullo, M.; Toledo, E.; Wang, D.D.; Corella, D.; Gomez-Gracia, E.; Fiol, M.; Estruch, R.; et al. Plasma Metabolites from Choline Pathway and Risk of Cardiovascular Disease in the PREDIMED (Prevention with Mediterranean Diet) Study. J. Am. Heart Assoc. 2017, 6, e006524.
- Millard, H.R.; Musani, S.K.; Dibaba, D.T.; Talegawkar, S.A.; Taylor, H.A.; Tucker, K.L.; Bidulescu, A. Dietary choline and betaine; associations with subclinical markers of cardiovascular disease risk and incidence of CVD, coronary heart disease and stroke: The Jackson Heart Study. Eur. J. Nutr. 2018, 57, 51–60.
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013, 2, e01202.
- Fazlollahi, M.; Chun, Y.; Grishin, A.; Wood, R.A.; Burks, A.W.; Dawson, P.; Jones, S.M.; Leung, D.Y.M.; Sampson, H.A.; Sicherer, S.H.; et al. Early-life gut microbiome and egg allergy. Allergy 2018, 73, 1515–1524.
- Bunyavanich, S.; Shen, N.; Grishin, A.; Wood, R.; Burks, W.; Dawson, P.; Jones, S.M.; Leung, D.Y.M.; Sampson, H.; Sicherer, S.; et al. Early-life gut microbiome composition and milk allergy resolution. J. Allergy Clin. Immunol. 2016, 138, 1122–1130.
- Osbelt, L.; Wende, M.; Almasi, E.; Derksen, E.; Muthukumarasamy, U.; Lesker, T.R.; Galvez, E.J.C.; Pils, M.C.; Schalk, E.; Chhatwal, P.; et al. Klebsiella oxytoca causes colonization resistance against multidrug-resistant K. pneumoniae in the gut via cooperative carbohydrate competition. Cell Host Microbe 2021, 29, 1663–1679.
- Osbelt, L.; Thiemann, S.; Smit, N.; Lesker, T.R.; Schroter, M.; Galvez, E.J.C.; Schmidt-Hohagen, K.; Pils, M.C.; Muhlen, S.; Dersch, P.; et al. Variations in microbiota composition of laboratory mice influence Citrobacter rodentium infection via variable short-chain fatty acid production. PLoS Pathog. 2020, 16, e1008448.
- Oliveira, R.A.; Ng, K.M.; Correia, M.B.; Cabral, V.; Shi, H.; Sonnenburg, J.L.; Huang, K.C.; Xavier, K.B. Klebsiella michiganensis transmission enhances resistance to Enterobacteriaceae gut invasion by nutrition competition. Nat. Microbiol. 2020, 5, 630–641.
- Ducarmon, Q.R.; Zwittink, R.D.; Hornung, B.V.H.; van Schaik, W.; Young, V.B.; Kuijper, E.J. Gut Microbiota and Colonization Resistance against Bacterial Enteric Infection. Microbiol. Mol. Biol. Rev. 2019, 83, e00007-19.
- Jacobson, A.; Lam, L.; Rajendram, M.; Tamburini, F.; Honeycutt, J.; Pham, T.; Van Treuren, W.; Pruss, K.; Stabler, S.R.; Lugo, K.; et al. A Gut Commensal-Produced Metabolite Mediates Colonization Resistance to Salmonella Infection. Cell Host Microbe 2018, 24, 296–307.
- Izquierdo, M.; Lopez, J.; Gallardo, P.; Vidal, R.M.; Ossa, J.C.; Farfan, M.J. Bacteria from gut microbiota associated with diarrheal infections in children promote virulence of Shiga toxin-producing and enteroaggregative Escherichia coli pathotypes. Front. Cell. Infect. Microbiol. 2022, 12, 867205.
- Lesniak, N.A.; Schubert, A.M.; Flynn, K.J.; Leslie, J.L.; Sinani, H.; Bergin, I.L.; Young, V.B.; Schloss, P.D. The Gut Bacterial Community Potentiates Clostridioides difficile Infection Severity. mBio 2022, 13, e0118322.
- Thiemann, S.; Smit, N.; Roy, U.; Lesker, T.R.; Galvez, E.J.C.; Helmecke, J.; Basic, M.; Bleich, A.; Goodman, A.L.; Kalinke, U.; et al. Enhancement of IFNgamma Production by Distinct Commensals Ameliorates Salmonella-Induced Disease. Cell Host Microbe 2017, 21, 682–694.e685.
- Caballero, S.; Kim, S.; Carter, R.A.; Leiner, I.M.; Susac, B.; Miller, L.; Kim, G.J.; Ling, L.; Pamer, E.G. Cooperating Commensals Restore Colonization Resistance to Vancomycin-Resistant Enterococcus faecium. Cell Host Microbe 2017, 21, 592–602.e594.
- Bibbo, S.; Lopetuso, L.R.; Ianiro, G.; Di Rienzo, T.; Gasbarrini, A.; Cammarota, G. Role of microbiota and innate immunity in recurrent Clostridium difficile infection. J. Immunol. Res. 2014, 2014, 462740.
- Warny, M.; Keates, A.C.; Keates, S.; Castagliuolo, I.; Zacks, J.K.; Aboudola, S.; Qamar, A.; Pothoulakis, C.; LaMont, J.T.; Kelly, C.P. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J. Clin. Investig. 2000, 105, 1147–1156.
- Ishida, Y.; Maegawa, T.; Kondo, T.; Kimura, A.; Iwakura, Y.; Nakamura, S.; Mukaida, N. Essential involvement of IFN-gamma in Clostridium difficile toxin A-induced enteritis. J. Immunol. 2004, 172, 3018–3025.
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E., Jr.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.Z.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114.
- Rea, M.C.; Dobson, A.; O′Sullivan, O.; Crispie, F.; Fouhy, F.; Cotter, P.D.; Shanahan, F.; Kiely, B.; Hill, C.; Ross, R.P. Effect of broad- and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4639–4644.
- Buffie, C.G.; Jarchum, I.; Equinda, M.; Lipuma, L.; Gobourne, A.; Viale, A.; Ubeda, C.; Xavier, J.; Pamer, E.G. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 2012, 80, 62–73.
- McDonald, L.C. Effects of short- and long-course antibiotics on the lower intestinal microbiome as they relate to traveller′s diarrhea. J. Travel Med. 2017, 24, S35–S38.
- Wang, S.; Deng, W.; Li, F.; Chen, Y.E.; Wang, P.U. Blockade of T helper 17 cell function ameliorates recurrent Clostridioides difficile infection in mice. Acta Biochim. Biophys. Sin. 2021, 53, 1290–1299.
- Frisbee, A.L.; Saleh, M.M.; Young, M.K.; Leslie, J.L.; Simpson, M.E.; Abhyankar, M.M.; Cowardin, C.A.; Ma, J.Z.; Pramoonjago, P.; Turner, S.D.; et al. IL-33 drives group 2 innate lymphoid cell-mediated protection during Clostridium difficile infection. Nat. Commun. 2019, 10, 2712.
- Wu, Z.; Xu, Q.; Wang, Q.; Chen, Y.; Lv, L.; Zheng, B.; Yan, R.; Jiang, H.; Shen, J.; Wang, S.; et al. The impact of dietary fibers on Clostridioides difficile infection in a mouse model. Front. Cell. Infect. Microbiol. 2022, 12, 1028267.
- Hayashi, A.; Nagao-Kitamoto, H.; Kitamoto, S.; Kim, C.H.; Kamada, N. The Butyrate-Producing Bacterium Clostridium butyricum Suppresses Clostridioides difficile Infection via Neutrophil- and Antimicrobial Cytokine-Dependent but GPR43/109a-Independent Mechanisms. J. Immunol. 2021, 206, 1576–1585.
- Green, J.E.; Davis, J.A.; Berk, M.; Hair, C.; Loughman, A.; Castle, D.; Athan, E.; Nierenberg, A.A.; Cryan, J.F.; Jacka, F.; et al. Efficacy and safety of fecal microbiota transplantation for the treatment of diseases other than Clostridium difficile infection: A systematic review and meta-analysis. Gut Microbes 2020, 12, 1854640.
- Petrof, E.O.; Gloor, G.B.; Vanner, S.J.; Weese, S.J.; Carter, D.; Daigneault, M.C.; Brown, E.M.; Schroeter, K.; Allen-Vercoe, E. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ′RePOOPulating′ the gut. Microbiome 2013, 1, 3.
- Martz, S.L.; McDonald, J.A.; Sun, J.; Zhang, Y.G.; Gloor, G.B.; Noordhof, C.; He, S.M.; Gerbaba, T.K.; Blennerhassett, M.; Hurlbut, D.J.; et al. Administration of defined microbiota is protective in a murine Salmonella infection model. Sci. Rep. 2015, 5, 16094.
- Barbara, G.; Barbaro, M.R.; Fuschi, D.; Palombo, M.; Falangone, F.; Cremon, C.; Marasco, G.; Stanghellini, V. Inflammatory and Microbiota-Related Regulation of the Intestinal Epithelial Barrier. Front. Nutr. 2021, 8, 718356.
- Chassaing, B.; Darfeuille-Michaud, A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011, 140, 1720–1728.
- Swidsinski, A.; Ladhoff, A.; Pernthaler, A.; Swidsinski, S.; Loening-Baucke, V.; Ortner, M.; Weber, J.; Hoffmann, U.; Schreiber, S.; Dietel, M.; et al. Mucosal flora in inflammatory bowel disease. Gastroenterology 2002, 122, 44–54.
- Schwartz, S.M.; Kemnitz, J.W. Age- and gender-related changes in body size, adiposity, and endocrine and metabolic parameters in free-ranging rhesus macaques. Am. J. Phys. Anthropol. 1992, 89, 109–121.
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vazquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn′s disease. Cell Host Microbe 2014, 15, 382–392.
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283.
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 2012, 487, 104–108.
- Shapiro, J.M.; de Zoete, M.R.; Palm, N.W.; Laenen, Y.; Bright, R.; Mallette, M.; Bu, K.; Bielecka, A.A.; Xu, F.; Hurtado-Lorenzo, A.; et al. Immunoglobulin A Targets a Unique Subset of the Microbiota in Inflammatory Bowel Disease. Cell Host Microbe 2021, 29, 83–93.
- Palm, N.W.; de Zoete, M.R.; Cullen, T.W.; Barry, N.A.; Stefanowski, J.; Hao, L.; Degnan, P.H.; Hu, J.; Peter, I.; Zhang, W.; et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014, 158, 1000–1010.
- Schaubeck, M.; Clavel, T.; Calasan, J.; Lagkouvardos, I.; Haange, S.B.; Jehmlich, N.; Basic, M.; Dupont, A.; Hornef, M.; von Bergen, M.; et al. Dysbiotic gut microbiota causes transmissible Crohn′s disease-like ileitis independent of failure in antimicrobial defence. Gut 2016, 65, 225–237.
- Torres, J.; Hu, J.; Seki, A.; Eisele, C.; Nair, N.; Huang, R.; Tarassishin, L.; Jharap, B.; Cote-Daigneault, J.; Mao, Q.; et al. Infants born to mothers with IBD present with altered gut microbiome that transfers abnormalities of the adaptive immune system to germ-free mice. Gut 2020, 69, 42–51.
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305.
- Huang, C.; Mei, Q.; Lou, L.; Huang, Z.; Fu, Y.; Fan, J.; Wang, J.; Yin, N.; Zheng, Y.; Lu, Y.; et al. Ulcerative Colitis in Response to Fecal Microbiota Transplantation via Modulation of Gut Microbiota and Th17/Treg Cell Balance. Cells 2022, 11, 1851.
- Facchin, S.; Vitulo, N.; Calgaro, M.; Buda, A.; Romualdi, C.; Pohl, D.; Perini, B.; Lorenzon, G.; Marinelli, C.; D′Inca, R.; et al. Microbiota changes induced by microencapsulated sodium butyrate in patients with inflammatory bowel disease. Neurogastroenterol. Motil. 2020, 32, e13914.
- Li, G.; Lin, J.; Zhang, C.; Gao, H.; Lu, H.; Gao, X.; Zhu, R.; Li, Z.; Li, M.; Liu, Z. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes 2021, 13, 1968257.
- Hartog, A.; Belle, F.N.; Bastiaans, J.; de Graaff, P.; Garssen, J.; Harthoorn, L.F.; Vos, A.P. A potential role for regulatory T-cells in the amelioration of DSS induced colitis by dietary non-digestible polysaccharides. J. Nutr. Biochem. 2015, 26, 227–233.
- Singh, V.; Yeoh, B.S.; Walker, R.E.; Xiao, X.; Saha, P.; Golonka, R.M.; Cai, J.; Bretin, A.C.A.; Cheng, X.; Liu, Q.; et al. Microbiota fermentation-NLRP3 axis shapes the impact of dietary fibres on intestinal inflammation. Gut 2019, 68, 1801–1812.
- Zhu, Y.; Xu, Y.; Wang, X.; Rao, L.; Yan, X.; Gao, R.; Shen, T.; Zhou, Y.; Kong, C.; Zhou, L. Probiotic Cocktail Alleviates Intestinal Inflammation Through Improving Gut Microbiota and Metabolites in Colitis Mice. Front. Cell. Infect. Microbiol. 2022, 12, 886061.
- Xu, M.; Shen, Y.; Cen, M.; Zhu, Y.; Cheng, F.; Tang, L.; Zheng, X.; Kim, J.J.; Dai, N.; Hu, W. Modulation of the Gut Microbiota-farnesoid X Receptor Axis Improves Deoxycholic Acid-induced Intestinal Inflammation in Mice. J. Crohn’s Colitis 2021, 15, 1197–1210.
- Xu, M.; Cen, M.; Shen, Y.; Zhu, Y.; Cheng, F.; Tang, L.; Hu, W.; Dai, N. Deoxycholic Acid-Induced Gut Dysbiosis Disrupts Bile Acid Enterohepatic Circulation and Promotes Intestinal Inflammation. Dig. Dis. Sci. 2021, 66, 568–576.
- Liu, T.C.; Kern, J.T.; Jain, U.; Sonnek, N.M.; Xiong, S.; Simpson, K.F.; VanDussen, K.L.; Winkler, E.S.; Haritunians, T.; Malique, A.; et al. Western diet induces Paneth cell defects through microbiome alterations and farnesoid X receptor and type I interferon activation. Cell Host Microbe 2021, 29, 988–1001.
- Liu, Y.; Xu, J.; Ren, X.; Zhang, Y.; Ke, Z.; Zhou, J.; Wang, Y.; Zhang, Y.; Liu, Y. Cholecystectomy-induced secondary bile acids accumulation ameliorates colitis through inhibiting monocyte/macrophage recruitment. Gut Microbes 2022, 14, 2107387.
- Van den Bossche, L.; Hindryckx, P.; Devisscher, L.; Devriese, S.; Van Welden, S.; Holvoet, T.; Vilchez-Vargas, R.; Vital, M.; Pieper, D.H.; Vanden Bussche, J.; et al. Ursodeoxycholic Acid and Its Taurine- or Glycine-Conjugated Species Reduce Colitogenic Dysbiosis and Equally Suppress Experimental Colitis in Mice. Appl. Environ. Microbiol. 2017, 83, e02766-16.
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539.
- Moltzau Anderson, J.; Lipinski, S.; Sommer, F.; Pan, W.H.; Boulard, O.; Rehman, A.; Falk-Paulsen, M.; Stengel, S.T.; Aden, K.; Hasler, R.; et al. NOD2 Influences Trajectories of Intestinal Microbiota Recovery After Antibiotic Perturbation. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 365–389.
- Danne, C.; Michaudel, C.; Skerniskyte, J.; Planchais, J.; Magniez, A.; Agus, A.; Michel, M.L.; Lamas, B.; Da Costa, G.; Spatz, M.; et al. CARD9 in neutrophils protects from colitis and controls mitochondrial metabolism and cell survival. Gut 2022.
- Lavoie, S.; Conway, K.L.; Lassen, K.G.; Jijon, H.B.; Pan, H.; Chun, E.; Michaud, M.; Lang, J.K.; Gallini Comeau, C.A.; Dreyfuss, J.M.; et al. The Crohn′s disease polymorphism, ATG16L1 T300A, alters the gut microbiota and enhances the local Th1/Th17 response. Elife 2019, 8, e39982.
- Aden, K.; Tran, F.; Ito, G.; Sheibani-Tezerji, R.; Lipinski, S.; Kuiper, J.W.; Tschurtschenthaler, M.; Saveljeva, S.; Bhattacharyya, J.; Hasler, R.; et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J. Exp. Med. 2018, 215, 2868–2886.
- Zhou, C.B.; Zhou, Y.L.; Fang, J.Y. Gut Microbiota in Cancer Immune Response and Immunotherapy. Trends Cancer 2021, 7, 647–660.
- Xu, C.; Fan, L.; Lin, Y.; Shen, W.; Qi, Y.; Zhang, Y.; Chen, Z.; Wang, L.; Long, Y.; Hou, T.; et al. Fusobacterium nucleatum promotes colorectal cancer metastasis through miR-1322/CCL20 axis and M2 polarization. Gut Microbes 2021, 13, 1980347.
- Hussan, H.; Clinton, S.K.; Roberts, K.; Bailey, M.T. Fusobacterium′s link to colorectal neoplasia sequenced: A systematic review and future insights. World J. Gastroenterol. 2017, 23, 8626–8650.
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-kappaB, and Up-regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–866.
- Gur, C.; Maalouf, N.; Shhadeh, A.; Berhani, O.; Singer, B.B.; Bachrach, G.; Mandelboim, O. Fusobacterium nucleatum supresses anti-tumor immunity by activating CEACAM1. Oncoimmunology 2019, 8, e1581531.
- Brennan, C.A.; Clay, S.L.; Lavoie, S.L.; Bae, S.; Lang, J.K.; Fonseca-Pereira, D.; Rosinski, K.G.; Ou, N.; Glickman, J.N.; Garrett, W.S. Fusobacterium nucleatum drives a pro-inflammatory intestinal microenvironment through metabolite receptor-dependent modulation of IL-17 expression. Gut Microbes 2021, 13, 1987780.
- Loesche, W.J.; Gibbons, R.J. Amino acid fermentation by Fusobacterium nucleatum. Arch. Oral Biol. 1968, 13, 191–202.
- Lavoie, S.; Chun, E.; Bae, S.; Brennan, C.A.; Gallini Comeau, C.A.; Lang, J.K.; Michaud, M.; Hoveyda, H.R.; Fraser, G.L.; Fuller, M.H.; et al. Expression of Free Fatty Acid Receptor 2 by Dendritic Cells Prevents Their Expression of Interleukin 27 and Is Required for Maintenance of Mucosal Barrier and Immune Response Against Colorectal Tumors in Mice. Gastroenterology 2020, 158, 1359–1372.
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014, 4, 1387–1397.
- Dong, W.; Liu, L.; Dou, Y.; Xu, M.; Liu, T.; Wang, S.; Zhang, Y.; Deng, B.; Wang, B.; Cao, H. Deoxycholic acid activates epidermal growth factor receptor and promotes intestinal carcinogenesis by ADAM17-dependent ligand release. J. Cell. Mol. Med. 2018, 22, 4263–4273.
- Pai, R.; Tarnawski, A.S.; Tran, T. Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol. Biol. Cell 2004, 15, 2156–2163.
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112.
- Liu, L.; Yang, M.; Dong, W.; Liu, T.; Song, X.; Gu, Y.; Wang, S.; Liu, Y.; Abla, Z.; Qiao, X.; et al. Gut Dysbiosis and Abnormal Bile Acid Metabolism in Colitis-Associated Cancer. Gastroenterol. Res. Pract. 2021, 2021, 6645970.
- Ternes, D.; Tsenkova, M.; Pozdeev, V.I.; Meyers, M.; Koncina, E.; Atatri, S.; Schmitz, M.; Karta, J.; Schmoetten, M.; Heinken, A.; et al. The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat. Metab. 2022, 4, 458–475.
- Fan, L.; Xu, C.; Ge, Q.; Lin, Y.; Wong, C.C.; Qi, Y.; Ye, B.; Lian, Q.; Zhuo, W.; Si, J.; et al. A. muciniphila Suppresses Colorectal Tumorigenesis by Inducing TLR2/NLRP3-Mediated M1-Like TAMs. Cancer Immunol. Res. 2021, 9, 1111–1124.
- Zegarra Ruiz, D.F.; Kim, D.V.; Norwood, K.; Saldana-Morales, F.B.; Kim, M.; Ng, C.; Callaghan, R.; Uddin, M.; Chang, L.C.; Longman, R.S.; et al. Microbiota manipulation to increase macrophage IL-10 improves colitis and limits colitis-associated colorectal cancer. Gut Microbes 2022, 14, 2119054.
- Yu, A.I.; Zhao, L.; Eaton, K.A.; Ho, S.; Chen, J.; Poe, S.; Becker, J.; Gonzalez, A.; McKinstry, D.; Hasso, M.; et al. Gut Microbiota Modulate CD8 T Cell Responses to Influence Colitis-Associated Tumorigenesis. Cell Rep. 2020, 31, 107471.
- Peuker, K.; Strigli, A.; Tauriello, D.V.F.; Hendricks, A.; von Schonfels, W.; Burmeister, G.; Brosch, M.; Herrmann, A.; Kruger, S.; Nitsche, J.; et al. Microbiota-dependent activation of the myeloid calcineurin-NFAT pathway inhibits B7H3- and B7H4-dependent anti-tumor immunity in colorectal cancer. Immunity 2022, 55, 701–717.
- Overacre-Delgoffe, A.E.; Bumgarner, H.J.; Cillo, A.R.; Burr, A.H.P.; Tometich, J.T.; Bhattacharjee, A.; Bruno, T.C.; Vignali, D.A.A.; Hand, T.W. Microbiota-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. Immunity 2021, 54, 2812–2824.e2814.
- Noble, A.; Pring, E.T.; Durant, L.; Man, R.; Dilke, S.M.; Hoyles, L.; James, S.A.; Carding, S.R.; Jenkins, J.T.; Knight, S.C. Altered immunity to microbiota, B cell activation and depleted gammadelta/resident memory T cells in colorectal cancer. Cancer Immunol. Immunother. 2022, 71, 2619–2629.
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604.
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238.
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208.
- Said, I.; Ahad, H.; Said, A. Gut microbiome in non-alcoholic fatty liver disease associated hepatocellular carcinoma: Current knowledge and potential for therapeutics. World J. Gastrointest. Oncol. 2022, 14, 947–958.
- Behary, J.; Raposo, A.E.; Amorim, N.M.L.; Zheng, H.; Gong, L.; McGovern, E.; Chen, J.; Liu, K.; Beretov, J.; Theocharous, C.; et al. Defining the temporal evolution of gut dysbiosis and inflammatory responses leading to hepatocellular carcinoma in Mdr2 −/− mouse model. BMC Microbiol. 2021, 21, 113.
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated with Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120.
- Zheng, R.; Wang, G.; Pang, Z.; Ran, N.; Gu, Y.; Guan, X.; Yuan, Y.; Zuo, X.; Pan, H.; Zheng, J.; et al. Liver cirrhosis contributes to the disorder of gut microbiota in patients with hepatocellular carcinoma. Cancer Med. 2020, 9, 4232–4250.
- Liu, Q.; Li, F.; Zhuang, Y.; Xu, J.; Wang, J.; Mao, X.; Zhang, Y.; Liu, X. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. 2019, 11, 1.
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023.
- Grat, M.; Wronka, K.M.; Krasnodebski, M.; Masior, L.; Lewandowski, Z.; Kosinska, I.; Grat, K.; Stypulkowski, J.; Rejowski, S.; Wasilewicz, M.; et al. Profile of Gut Microbiota Associated with the Presence of Hepatocellular Cancer in Patients with Liver Cirrhosis. Transplant. Proc. 2016, 48, 1687–1691.
- Ohtani, N.; Hara, E. Gut-liver axis-mediated mechanism of liver cancer: A special focus on the role of gut microbiota. Cancer Sci. 2021, 112, 4433–4443.
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE(2)-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538.
- Golonka, R.M.; Xiao, X.; Abokor, A.A.; Joe, B.; Vijay-Kumar, M. Altered nutrient status reprograms host inflammation and metabolic health via gut microbiota. J. Nutr. Biochem. 2020, 80, 108360.
- Hosseinkhani, F.; Heinken, A.; Thiele, I.; Lindenburg, P.W.; Harms, A.C.; Hankemeier, T. The contribution of gut bacterial metabolites in the human immune signaling pathway of non-communicable diseases. Gut Microbes 2021, 13, 1882927.
- Yue, C.; Yang, X.; Li, J.; Chen, X.; Zhao, X.; Chen, Y.; Wen, Y. Trimethylamine N-oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem. Biophys. Res. Commun. 2017, 490, 541–551.
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63.
- Guasti, L.; Galliazzo, S.; Molaro, M.; Visconti, E.; Pennella, B.; Gaudio, G.V.; Lupi, A.; Grandi, A.M.; Squizzato, A. TMAO as a biomarker of cardiovascular events: A systematic review and meta-analysis. Intern. Emerg. Med. 2021, 16, 201–207.
- Fang, C.; Zuo, K.; Jiao, K.; Zhu, X.; Fu, Y.; Zhong, J.; Xu, L.; Yang, X. PAGln, an Atrial Fibrillation-Linked Gut Microbial Metabolite, Acts as a Promoter of Atrial Myocyte Injury. Biomolecules 2022, 12, 1120.
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.
- Bell, K.J.; Saad, S.; Tillett, B.J.; McGuire, H.M.; Bordbar, S.; Yap, Y.A.; Nguyen, L.T.; Wilkins, M.R.; Corley, S.; Brodie, S.; et al. Metabolite-based dietary supplementation in human type 1 diabetes is associated with microbiota and immune modulation. Microbiome 2022, 10, 9.
- Marino, E.; Richards, J.L.; McLeod, K.H.; Stanley, D.; Yap, Y.A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J.; et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 2017, 18, 552–562.
- Girdhar, K.; Huang, Q.; Chow, I.T.; Vatanen, T.; Brady, C.; Raisingani, A.; Autissier, P.; Atkinson, M.A.; Kwok, W.W.; Kahn, C.R.; et al. A gut microbial peptide and molecular mimicry in the pathogenesis of type 1 diabetes. Proc. Natl. Acad. Sci. USA 2022, 119, e2120028119.
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Alves, M.A.; Harkonen, T.; Honkanen, J.; Vatanen, T.; Xavier, R.J.; Hyotylainen, T.; Knip, M.; et al. Dysregulation of secondary bile acid metabolism precedes islet autoimmunity and type 1 diabetes. Cell Rep. Med. 2022, 3, 100762.
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381.
- Zhou, M.; Shao, J.; Wu, C.Y.; Shu, L.; Dong, W.; Liu, Y.; Chen, M.; Wynn, R.M.; Wang, J.; Wang, J.; et al. Targeting BCAA Catabolism to Treat Obesity-Associated Insulin Resistance. Diabetes 2019, 68, 1730–1746.
- Bodogai, M.; O′Connell, J.; Kim, K.; Kim, Y.; Moritoh, K.; Chen, C.; Gusev, F.; Vaughan, K.; Shulzhenko, N.; Mattison, J.A.; et al. Commensal bacteria contribute to insulin resistance in aging by activating innate B1a cells. Sci. Transl. Med. 2018, 10, eaat4271.
- Huang, J.P.; Cheng, M.L.; Hung, C.Y.; Wang, C.H.; Hsieh, P.S.; Shiao, M.S.; Chen, J.K.; Li, D.E.; Hung, L.M. Docosapentaenoic acid and docosahexaenoic acid are positively associated with insulin sensitivity in rats fed high-fat and high-fructose diets. J. Diabetes 2017, 9, 936–946.
- Wang, H.; Lu, Y.; Yan, Y.; Tian, S.; Zheng, D.; Leng, D.; Wang, C.; Jiao, J.; Wang, Z.; Bai, Y. Promising Treatment for Type 2 Diabetes: Fecal Microbiota Transplantation Reverses Insulin Resistance and Impaired Islets. Front. Cell. Infect. Microbiol. 2019, 9, 455.
- Deng, X.; Zhang, C.; Wang, P.; Wei, W.; Shi, X.; Wang, P.; Yang, J.; Wang, L.; Tang, S.; Fang, Y.; et al. Cardiovascular Benefits of Empagliflozin Are Associated with Gut Microbiota and Plasma Metabolites in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2022, 107, 1888–1896.
- Montandon, S.A.; Jornayvaz, F.R. Effects of Antidiabetic Drugs on Gut Microbiota Composition. Genes 2017, 8, 250.
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858.
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14.
- Adnan, S.; Nelson, J.W.; Ajami, N.J.; Venna, V.R.; Petrosino, J.F.; Bryan, R.M., Jr.; Durgan, D.J. Alterations in the gut microbiota can elicit hypertension in rats. Physiol. Genom. 2017, 49, 96–104.
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977.
- Santisteban, M.M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.O.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ. Res. 2017, 120, 312–323.
- Karbach, S.H.; Schonfelder, T.; Brandao, I.; Wilms, E.; Hormann, N.; Jackel, S.; Schuler, R.; Finger, S.; Knorr, M.; Lagrange, J.; et al. Gut Microbiota Promote Angiotensin II-Induced Arterial Hypertension and Vascular Dysfunction. J. Am. Heart Assoc. 2016, 5, e003698.
- Avery, E.G.; Bartolomaeus, H.; Rauch, A.; Chen, C.Y.; N′Diaye, G.; Lober, U.; Bartolomaeus, T.U.P.; Fritsche-Guenther, R.; Rodrigues, A.F.; Yarritu, A.; et al. Quantifying the impact of gut microbiota on inflammation and hypertensive organ damage. Cardiovasc. Res. 2022.
- Joe, B.; McCarthy, C.G.; Edwards, J.M.; Cheng, X.; Chakraborty, S.; Yang, T.; Golonka, R.M.; Mell, B.; Yeo, J.Y.; Bearss, N.R.; et al. Microbiota Introduced to Germ-Free Rats Restores Vascular Contractility and Blood Pressure. Hypertension 2020, 76, 1847–1855.
- Abboud, F.M.; Cicha, M.Z.; Ericsson, A.; Chapleau, M.W.; Singh, M.V. Altering Early Life Gut Microbiota Has Long-Term Effect on Immune System and Hypertension in Spontaneously Hypertensive Rats. Front. Physiol. 2021, 12, 752924.
- Jiang, S.; Shui, Y.; Cui, Y.; Tang, C.; Wang, X.; Qiu, X.; Hu, W.; Fei, L.; Li, Y.; Zhang, S.; et al. Gut microbiota dependent trimethylamine N-oxide aggravates angiotensin II-induced hypertension. Redox Biol. 2021, 46, 102115.
- Ferguson, J.F.; Aden, L.A.; Barbaro, N.R.; Van Beusecum, J.P.; Xiao, L.; Simmons, A.J.; Warden, C.; Pasic, L.; Himmel, L.E.; Washington, M.K.; et al. High dietary salt-induced dendritic cell activation underlies microbial dysbiosis-associated hypertension. JCI Insight 2019, 5, e126241.
- Chakraborty, S.; Galla, S.; Cheng, X.; Yeo, J.Y.; Mell, B.; Singh, V.; Yeoh, B.; Saha, P.; Mathew, A.V.; Vijay-Kumar, M.; et al. Salt-Responsive Metabolite, beta-Hydroxybutyrate, Attenuates Hypertension. Cell Rep. 2018, 25, 677–689.
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mahler, A.; Balogh, A.; Marko, L.; et al. Salt-responsive gut commensal modulates T(H)17 axis and disease. Nature 2017, 551, 585–589.
- Lee, Y.H. Causal association of gut microbiome on the risk of rheumatoid arthritis: A Mendelian randomisation study. Ann. Rheum. Dis. 2022, 81, e3.
- Kishikawa, T.; Maeda, Y.; Nii, T.; Motooka, D.; Matsumoto, Y.; Matsushita, M.; Matsuoka, H.; Yoshimura, M.; Kawada, S.; Teshigawara, S.; et al. Metagenome-wide association study of gut microbiome revealed novel aetiology of rheumatoid arthritis in the Japanese population. Ann. Rheum. Dis. 2020, 79, 103–111.
- Pianta, A.; Arvikar, S.; Strle, K.; Drouin, E.E.; Wang, Q.; Costello, C.E.; Steere, A.C. Evidence of the Immune Relevance of Prevotella copri, a Gut Microbe, in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2017, 69, 964–975.
- Li, Y.; Zhang, S.X.; Yin, X.F.; Zhang, M.X.; Qiao, J.; Xin, X.H.; Chang, M.J.; Gao, C.; Li, Y.F.; Li, X.F. The Gut Microbiota and Its Relevance to Peripheral Lymphocyte Subpopulations and Cytokines in Patients with Rheumatoid Arthritis. J. Immunol. Res. 2021, 2021, 6665563.
- Teng, F.; Klinger, C.N.; Felix, K.M.; Bradley, C.P.; Wu, E.; Tran, N.L.; Umesaki, Y.; Wu, H.J. Gut Microbiota Drive Autoimmune Arthritis by Promoting Differentiation and Migration of Peyer′s Patch T Follicular Helper Cells. Immunity 2016, 44, 875–888.
- Bates, N.A.; Li, A.; Fan, T.; Cutcliffe, M.P.; Dagenet, C.B.; Sleiman, K.C.; Ma, H.; Tahsin, S.; Garrett, C.S.; Altemus, J.; et al. Gut Commensal Segmented Filamentous Bacteria Fine-Tune T Follicular Regulatory Cells to Modify the Severity of Systemic Autoimmune Arthritis. J. Immunol. 2021, 206, 941–952.
- Teng, F.; Felix, K.M.; Bradley, C.P.; Naskar, D.; Ma, H.; Raslan, W.A.; Wu, H.J. The impact of age and gut microbiota on Th17 and Tfh cells in K/BxN autoimmune arthritis. Arthritis Res. Ther. 2017, 19, 188.
- Block, K.E.; Zheng, Z.; Dent, A.L.; Kee, B.L.; Huang, H. Gut Microbiota Regulates K/BxN Autoimmune Arthritis through Follicular Helper T but Not Th17 Cells. J. Immunol. 2016, 196, 1550–1557.
- Simpkins, D.A.; Downton, P.; Gray, K.J.; Dickson, S.H.; Maidstone, R.J.; Konkel, J.E.; Hepworth, M.R.; Ray, D.W.; Bechtold, D.A.; Gibbs, J.E. Consequences of collagen induced inflammatory arthritis on circadian regulation of the gut microbiome. FASEB J. 2023, 37, e22704.
- He, J.; Chu, Y.; Li, J.; Meng, Q.; Liu, Y.; Jin, J.; Wang, Y.; Wang, J.; Huang, B.; Shi, L.; et al. Intestinal butyrate-metabolizing species contribute to autoantibody production and bone erosion in rheumatoid arthritis. Sci. Adv. 2022, 8, eabm1511.
- Takahashi, D.; Hoshina, N.; Kabumoto, Y.; Maeda, Y.; Suzuki, A.; Tanabe, H.; Isobe, J.; Yamada, T.; Muroi, K.; Yanagisawa, Y.; et al. Microbiota-derived butyrate limits the autoimmune response by promoting the differentiation of follicular regulatory T cells. EBioMedicine 2020, 58, 102913.
- Rosser, E.C.; Piper, C.J.M.; Matei, D.E.; Blair, P.A.; Rendeiro, A.F.; Orford, M.; Alber, D.G.; Krausgruber, T.; Catalan, D.; Klein, N.; et al. Microbiota-Derived Metabolites Suppress Arthritis by Amplifying Aryl-Hydrocarbon Receptor Activation in Regulatory B Cells. Cell Metab. 2020, 31, 837–851.
- Sun, H.; Guo, Y.; Wang, H.; Yin, A.; Hu, J.; Yuan, T.; Zhou, S.; Xu, W.; Wei, P.; Yin, S.; et al. Gut commensal Parabacteroides distasonis alleviates inflammatory arthritis. Gut 2023.
- Adeyeye, T.E.; Yeung, E.H.; McLain, A.C.; Lin, S.; Lawrence, D.A.; Bell, E.M. Wheeze and Food Allergies in Children Born via Cesarean Delivery: The Upstate KIDS Study. Am. J. Epidemiol. 2019, 188, 355–362.
- Loh, W.; Tang, M.L.K. The Epidemiology of Food Allergy in the Global Context. Int J. Environ. Res. Public Health 2018, 15, 2043.
- Leung, A.S.Y.; Wong, G.W.K.; Tang, M.L.K. Food allergy in the developing world. J. Allergy Clin. Immunol. 2018, 141, 76–78.
- Borbet, T.C.; Pawline, M.B.; Zhang, X.; Wipperman, M.F.; Reuter, S.; Maher, T.; Li, J.; Iizumi, T.; Gao, Z.; Daniele, M.; et al. Influence of the early-life gut microbiota on the immune responses to an inhaled allergen. Mucosal Immunol. 2022, 15, 1000–1011.
- Zhang, Q.; Cheng, L.; Wang, J.; Hao, M.; Che, H. Antibiotic-Induced Gut Microbiota Dysbiosis Damages the Intestinal Barrier, Increasing Food Allergy in Adult Mice. Nutrients 2021, 13, 3315.
- Hussain, M.; Bonilla-Rosso, G.; Kwong Chung, C.K.C.; Bariswyl, L.; Rodriguez, M.P.; Kim, B.S.; Engel, P.; Noti, M. High dietary fat intake induces a microbiota signature that promotes food allergy. J. Allergy Clin. Immunol. 2019, 144, 157–170.
- Zimmermann, P.; Messina, N.; Mohn, W.W.; Finlay, B.B.; Curtis, N. Association between the intestinal microbiota and allergic sensitization, eczema, and asthma: A systematic review. J. Allergy Clin. Immunol. 2019, 143, 467–485.
- McCauley, K.E.; Rackaityte, E.; LaMere, B.; Fadrosh, D.W.; Fujimura, K.E.; Panzer, A.R.; Lin, D.L.; Lynch, K.V.; Halkias, J.; Mendoza, V.F.; et al. Heritable vaginal bacteria influence immune tolerance and relate to early-life markers of allergic sensitization in infancy. Cell Rep. Med. 2022, 3, 100713.
- Okba, A.M.; Saber, S.M.; Abdel-Rehim, A.S.; Amin, M.M.; Mohamed, D.A. Fecal microbiota profile in atopic asthmatic adult patients. Eur. Ann. Allergy Clin. Immunol. 2018, 50, 117–124.
- Lundstrom, S.L.; Yang, J.; Kallberg, H.J.; Thunberg, S.; Gafvelin, G.; Haeggstrom, J.Z.; Gronneberg, R.; Grunewald, J.; van Hage, M.; Hammock, B.D.; et al. Allergic asthmatics show divergent lipid mediator profiles from healthy controls both at baseline and following birch pollen provocation. PLoS ONE 2012, 7, e33780.
- Fujimura, K.E.; Sitarik, A.R.; Havstad, S.; Lin, D.L.; Levan, S.; Fadrosh, D.; Panzer, A.R.; LaMere, B.; Rackaityte, E.; Lukacs, N.W.; et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat. Med. 2016, 22, 1187–1191.
- Levan, S.R.; Stamnes, K.A.; Lin, D.L.; Panzer, A.R.; Fukui, E.; McCauley, K.; Fujimura, K.E.; McKean, M.; Ownby, D.R.; Zoratti, E.M.; et al. Elevated faecal 12,13-diHOME concentration in neonates at high risk for asthma is produced by gut bacteria and impedes immune tolerance. Nat. Microbiol. 2019, 4, 1851–1861.
- Abdulla, O.A.; Neamah, W.; Sultan, M.; Alghetaa, H.K.; Singh, N.; Busbee, P.B.; Nagarkatti, M.; Nagarkatti, P. The Ability of AhR Ligands to Attenuate Delayed Type Hypersensitivity Reaction Is Associated with Alterations in the Gut Microbiota. Front. Immunol. 2021, 12, 684727.
- Roduit, C.; Frei, R.; Ferstl, R.; Loeliger, S.; Westermann, P.; Rhyner, C.; Schiavi, E.; Barcik, W.; Rodriguez-Perez, N.; Wawrzyniak, M.; et al. High levels of butyrate and propionate in early life are associated with protection against atopy. Allergy 2019, 74, 799–809.
- Cait, A.; Hughes, M.R.; Antignano, F.; Cait, J.; Dimitriu, P.A.; Maas, K.R.; Reynolds, L.A.; Hacker, L.; Mohr, J.; Finlay, B.B.; et al. Microbiome-driven allergic lung inflammation is ameliorated by short-chain fatty acids. Mucosal Immunol. 2018, 11, 785–795.
- Thorburn, A.N.; McKenzie, C.I.; Shen, S.; Stanley, D.; Macia, L.; Mason, L.J.; Roberts, L.K.; Wong, C.H.; Shim, R.; Robert, R.; et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat. Commun. 2015, 6, 7320.
- Tan, J.; McKenzie, C.; Vuillermin, P.J.; Goverse, G.; Vinuesa, C.G.; Mebius, R.E.; Macia, L.; Mackay, C.R. Dietary Fiber and Bacterial SCFA Enhance Oral Tolerance and Protect against Food Allergy through Diverse Cellular Pathways. Cell Rep. 2016, 15, 2809–2824.
- Arifuzzaman, M.; Won, T.H.; Li, T.T.; Yano, H.; Digumarthi, S.; Heras, A.F.; Zhang, W.; Parkhurst, C.N.; Kashyap, S.; Jin, W.B.; et al. Inulin fibre promotes microbiota-derived bile acids and type 2 inflammation. Nature 2022, 611, 578–584.
- Seki, D.; Mayer, M.; Hausmann, B.; Pjevac, P.; Giordano, V.; Goeral, K.; Unterasinger, L.; Klebermass-Schrehof, K.; De Paepe, K.; Van de Wiele, T.; et al. Aberrant gut-microbiota-immune-brain axis development in premature neonates with brain damage. Cell Host Microbe 2021, 29, 1558–1572.
- Giuffre, M.; Moretti, R.; Campisciano, G.; da Silveira, A.B.M.; Monda, V.M.; Comar, M.; Di Bella, S.; Antonello, R.M.; Luzzati, R.; Croce, L.S. You Talking to Me? Says the Enteric Nervous System (ENS) to the Microbe. How Intestinal Microbes Interact with the ENS. J. Clin. Med. 2020, 9, 3705.
- Ouabbou, S.; He, Y.; Butler, K.; Tsuang, M. Inflammation in Mental Disorders: Is the Microbiota the Missing Link? Neurosci. Bull. 2020, 36, 1071–1084.
- Slyepchenko, A.; Carvalho, A.F.; Cha, D.S.; Kasper, S.; McIntyre, R.S. Gut emotions—Mechanisms of action of probiotics as novel therapeutic targets for depression and anxiety disorders. CNS Neurol. Disord. Drug Targets 2014, 13, 1770–1786.
- Zhang, X.; Hou, Y.; Li, Y.; Wei, W.; Cai, X.; Shao, H.; Yuan, Y.; Zheng, X. Taxonomic and Metabolic Signatures of Gut Microbiota for Assessing the Severity of Depression and Anxiety in Major Depressive Disorder Patients. Neuroscience 2022, 496, 179–189.
- Galley, J.D.; Mashburn-Warren, L.; Blalock, L.C.; Lauber, C.L.; Carroll, J.E.; Ross, K.M.; Hobel, C.; Coussons-Read, M.; Dunkel Schetter, C.; Gur, T.L. Maternal anxiety, depression and stress affects offspring gut microbiome diversity and bifidobacterial abundances. Brain Behav. Immun. 2023, 107, 253–264.
- O′Neill, S.M.; Curran, E.A.; Dalman, C.; Kenny, L.C.; Kearney, P.M.; Clarke, G.; Cryan, J.F.; Dinan, T.G.; Khashan, A.S. Birth by Caesarean Section and the Risk of Adult Psychosis: A Population-Based Cohort Study. Schizophr. Bull. 2016, 42, 633–641.
- Morais, L.H.; Golubeva, A.V.; Casey, S.; Scott, K.A.; Ramos Costa, A.P.; Moloney, G.M.; Dinan, T.G.; Cryan, J.F. Early-life oxytocin attenuates the social deficits induced by caesarean-section delivery in the mouse. Neuropsychopharmacology 2021, 46, 1958–1968.
- Lynch, C.M.K.; Cowan, C.S.M.; Bastiaanssen, T.F.S.; Moloney, G.M.; Theune, N.; van de Wouw, M.; Florensa Zanuy, E.; Ventura-Silva, A.P.; Codagnone, M.G.; Villalobos-Manriquez, F.; et al. Critical windows of early-life microbiota disruption on behaviour, neuroimmune function, and neurodevelopment. Brain Behav. Immun. 2022, 108, 309–327.
- Li, J.; Pu, F.; Peng, C.; Wang, Y.; Zhang, Y.; Wu, S.; Wang, S.; Shen, X.; Li, Y.; Cheng, R.; et al. Antibiotic cocktail-induced gut microbiota depletion in different stages could cause host cognitive impairment and emotional disorders in adulthood in different manners. Neurobiol. Dis. 2022, 170, 105757.
- Cuskelly, A.; Hoedt, E.C.; Harms, L.; Talley, N.J.; Tadros, M.A.; Keely, S.; Hodgson, D.M. Neonatal immune challenge influences the microbiota and behaviour in a sexually dimorphic manner. Brain Behav. Immun. 2022, 103, 232–242.
- Lee, J.; Venna, V.R.; Durgan, D.J.; Shi, H.; Hudobenko, J.; Putluri, N.; Petrosino, J.; McCullough, L.D.; Bryan, R.M. Young versus aged microbiota transplants to germ-free mice: Increased short-chain fatty acids and improved cognitive performance. Gut Microbes 2020, 12, 1814107.
- Leonardi, I.; Gao, I.H.; Lin, W.Y.; Allen, M.; Li, X.V.; Fiers, W.D.; De Celie, M.B.; Putzel, G.G.; Yantiss, R.K.; Johncilla, M.; et al. Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell 2022, 185, 831–846.
More
Information
Subjects:
Immunology; Microbiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.1K
Revisions:
2 times
(View History)
Update Date:
06 Feb 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No