 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mudit Tyagi | -- | 2407 | 2023-02-03 17:04:32 | | | |
| 2 | Jason Zhu | -1 word(s) | 2406 | 2023-02-06 06:44:28 | | |
Video Upload Options
The central nervous system (CNS) is highly compartmentalized and serves as a specific site of human immunodeficiency virus (HIV) infection. Therefore, an understanding of the cellular populations that are infected by HIV or that harbor latent HIV proviruses is imperative in the attempts to address cure strategies, taking into account that HIV infection and latency in the CNS may differ considerably from those in the periphery. HIV replication in the CNS is reported to persist despite prolonged combination antiretroviral therapy due to the inability of the antiretroviral drugs to penetrate and cross the blood–brain barrier.
1. Introduction
Human immunodeficiency virus type-1 (HIV-1) remains one of the most serious public health challenges [1][2][3][4]. HIV-1 invades the brain soon following systemic infection. Several mechanisms have been suggested for HIV-1 entry into the central nervous system (CNS). However, the “Trojan horse hypothesis”, which states that HIV-1 infection of the brain occurs through migration of the infected cells across the blood–brain barrier (BBB), is the most favored. Although CD4+ T cells, as well as monocytes and macrophages, are the primary cellular targets for productive HIV-1 infection [5][6][7], in the brain, macrophages and microglia are the main cell types productively infected by HIV-1, and virus production in the CNS is not seen before the onset of acquired immunodeficiency syndrome (AIDS) [8][9].
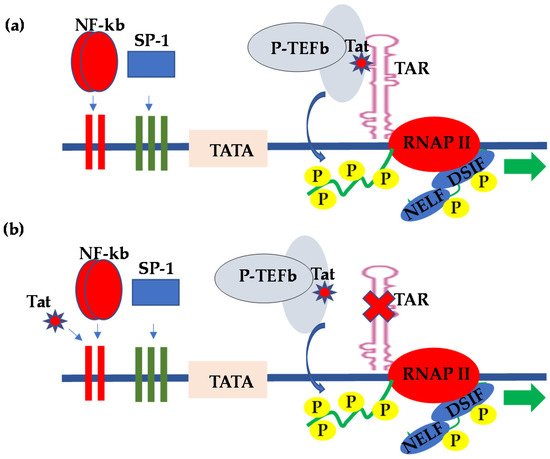
2. HIV Transcriptional Regulation in the CNS
| Anatomic Sites | Specific Sites |
|---|---|
| Brain | |
| Primary Lymphatic tissue/organ | Thymus and Bone marrow |
| Non-Lymphoid tissues | Liver, Kidney, Adipose, reproductive tract and other |
| Secondary lymphatic tissue/organs | Tonsils, Adenoids, spleen, Mucosa-associated lymphatic tissues and lymph nodes |
| Peripheral Blood |
3. HIV Latency in the CNS
References
- Chun, T.W.; Davey, R.T., Jr.; Engel, D.; Lane, H.C.; Fauci, A.S. Re-emergence of HIV after stopping therapy. Nature 1999, 401, 874–875.
- Hokello, J.; Sharma, A.L.; Tyagi, M. An Update on the HIV DNA Vaccine Strategy. Vaccines 2021, 9, 605.
- Stevenson, M. HIV-1 pathogenesis. Nat. Med. 2003, 9, 853–860.
- Hokello, J.; Sharma, A.L.; Tyagi, M. Efficient Non-Epigenetic Activation of HIV Latency through the T-Cell Receptor Signalosome. Viruses 2020, 12, 868.
- Fauci, A.S. Immunopathogenesis of HIV infection. J. Acquir. Immune Defic. Syndr. 1993, 6, 655–662.
- Poli, G.; Pantaleo, G.; Fauci, A.S. Immunopathogenesis of human immunodeficiency virus infection. Clin. Infect. Dis. 1993, 17 (Suppl. 1), S224–S229.
- Fauci, A.S. Multifactorial nature of human immunodeficiency virus disease: Implications for therapy. Science 1993, 262, 1011–1018.
- Kramer-Hammerle, S.; Rothenaigner, I.; Wolff, H.; Bell, J.E.; Brack-Werner, R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005, 111, 194–213.
- Cosenza, M.A.; Zhao, M.L.; Si, Q.; Lee, S.C. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 2002, 12, 442–455.
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25.
- Kingsman, S.M.; Kingsman, A.J. The regulation of human immunodeficiency virus type-1 gene expression. Eur. J. Biochem. 1996, 240, 491–507.
- Freed, E.O. HIV-1 replication. Somat. Cell Mol. Genet. 2001, 26, 13–33.
- Kim, Y.K.; Bourgeois, C.F.; Pearson, R.; Tyagi, M.; West, M.J.; Wong, J.; Wu, S.Y.; Chiang, C.M.; Karn, J. Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. EMBO J. 2006, 25, 3596–3604.
- Kumar, K.P.; Akoulitchev, S.; Reinberg, D. Promoter-proximal stalling results from the inability to recruit transcription factor IIH to the transcription complex and is a regulated event. Proc. Natl. Acad. Sci. USA 1998, 95, 9767–9772.
- Karn, J. Tackling Tat. J. Mol. Biol. 1999, 293, 235–254.
- Kim, Y.K.; Bourgeois, C.F.; Isel, C.; Churcher, M.J.; Karn, J. Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol. Cell. Biol. 2002, 22, 4622–4637.
- Marciniak, R.A.; Sharp, P.A. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 1991, 10, 4189–4196.
- Tyagi, M.; Rusnati, M.; Presta, M.; Giacca, M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2001, 276, 3254–3261.
- Schulze-Gahmen, U.; Hurley, J.H. Structural mechanism for HIV-1 TAR loop recognition by Tat and the super elongation complex. Proc. Natl. Acad. Sci. USA 2018, 115, 12973–12978.
- Hokello, J.; Sharma, A.L.; Tyagi, M. AP-1 and NF-kappaB synergize to transcriptionally activate latent HIV upon T-cell receptor activation. FEBS Lett. 2021, 595, 577–594.
- Yang, L.; Morris, G.F.; Lockyer, J.M.; Lu, M.; Wang, Z.; Morris, C.B. Distinct transcriptional pathways of TAR-dependent and TAR-independent human immunodeficiency virus type-1 transactivation by Tat. Virology 1997, 235, 48–64.
- Taylor, J.P.; Pomerantz, R.; Bagasra, O.; Chowdhury, M.; Rappaport, J.; Khalili, K.; Amini, S. TAR-independent transactivation by Tat in cells derived from the CNS: A novel mechanism of HIV-1 gene regulation. EMBO J. 1992, 11, 3395–3403.
- Taylor, J.P.; Pomerantz, R.J.; Oakes, J.W.; Khalili, K.; Amini, S. A CNS-enriched factor that binds to NF-kappa B and is required for interaction with HIV-1 tat. Oncogene 1995, 10, 395–400.
- Kundu, M.; Ansari, S.A.; Chepenik, L.G.; Pomerantz, R.J.; Khalili, K.; Rappaport, J.; Amini, S. HIV-1 regulatory protein tat induces RNA binding proteins in central nervous system cells that associate with the viral trans-acting-response regulatory motif. J. Hum. Virol. 1999, 2, 72–80.
- Taylor, J.P.; Pomerantz, R.J.; Raj, G.V.; Kashanchi, F.; Brady, J.N.; Amini, S.; Khalili, K. Central nervous system-derived cells express a kappa B-binding activity that enhances human immunodeficiency virus type 1 transcription in vitro and facilitates TAR-independent transactivation by Tat. J. Virol. 1994, 68, 3971–3981.
- Cupp, C.; Taylor, J.P.; Khalili, K.; Amini, S. Evidence for stimulation of the transforming growth factor beta 1 promoter by HIV-1 Tat in cells derived from CNS. Oncogene 1993, 8, 2231–2236.
- Sawaya, B.E.; Thatikunta, P.; Denisova, L.; Brady, J.; Khalili, K.; Amini, S. Regulation of TNFalpha and TGFbeta-1 gene transcription by HIV-1 Tat in CNS cells. J. Neuroimmunol. 1998, 87, 33–42.
- Gray, L.R.; Cowley, D.; Welsh, C.; Lu, H.K.; Brew, B.J.; Lewin, S.R.; Wesselingh, S.L.; Gorry, P.R.; Churchill, M.J. CNS-specific regulatory elements in brain-derived HIV-1 strains affect responses to latency-reversing agents with implications for cure strategies. Mol. Psychiatry 2016, 21, 574–584.
- Tyagi, M.; Bukrinsky, M.; Simon, G.L. Mechanisms of HIV Transcriptional Regulation by Drugs of Abuse. Curr. HIV Res. 2016, 14, 442–454.
- Tyagi, M.; Weber, J.; Bukrinsky, M.; Simon, G.L. The effects of cocaine on HIV transcription. J. Neurovirol. 2016, 22, 261–274.
- Swepson, C.; Ranjan, A.; Balasubramaniam, M.; Pandhare, J.; Dash, C. Cocaine Enhances HIV-1 Transcription in Macrophages by Inducing p38 MAPK Phosphorylation. Front. Microbiol. 2016, 7, 823.
- Reynolds, J.L.; Mahajan, S.D.; Bindukumar, B.; Sykes, D.; Schwartz, S.A.; Nair, M.P. Proteomic analysis of the effects of cocaine on the enhancement of HIV-1 replication in normal human astrocytes (NHA). Brain Res. 2006, 1123, 226–236.
- Sahu, G.; Farley, K.; El-Hage, N.; Aiamkitsumrit, B.; Fassnacht, R.; Kashanchi, F.; Ochem, A.; Simon, G.L.; Karn, J.; Hauser, K.F.; et al. Cocaine promotes both initiation and elongation phase of HIV-1 transcription by activating NF-kappaB and MSK1 and inducing selective epigenetic modifications at HIV-1 LTR. Virology 2015, 483, 185–202.
- Sonti, S.; Sharma, A.L.; Tyagi, M. HIV-1 persistence in the CNS: Mechanisms of latency, pathogenesis and an update on eradication strategies. Virus Res. 2021, 303, 198523.
- Sharma, A.L.; Hokello, J.; Sonti, S.; Zicari, S.; Sun, L.; Alqatawni, A.; Bukrinsky, M.; Simon, G.; Chauhan, A.; Daniel, R.; et al. CBF-1 Promotes the Establishment and Maintenance of HIV Latency by Recruiting Polycomb Repressive Complexes, PRC1 and PRC2, at HIV LTR. Viruses 2020, 12, 1040.
- Sharma, A.L.; Hokello, J.; Tyagi, M. Circumcision as an Intervening Strategy against HIV Acquisition in the Male Genital Tract. Pathogens 2021, 10, 806.
- Tyagi, M.; Bukrinsky, M. Human immunodeficiency virus (HIV) latency: The major hurdle in HIV eradication. Mol. Med. 2012, 18, 1096–1108.
- Zicari, S.; Sharma, A.L.; Sahu, G.; Dubrovsky, L.; Sun, L.; Yue, H.; Jada, T.; Ochem, A.; Simon, G.; Bukrinsky, M.; et al. DNA dependent protein kinase (DNA-PK) enhances HIV transcription by promoting RNA polymerase II activity and recruitment of transcription machinery at HIV LTR. Oncotarget 2020, 11, 699–726.
- Alqatawni, A.; Sharma, A.L.; Attilus, B.; Tyagi, M.; Daniel, R. Shedding Light on the Role of Extracellular Vesicles in HIV Infection and Wound Healing. Viruses 2020, 12, 584.
- Hokello, J.; Sharma, A.L.; Dimri, M.; Tyagi, M. Insights into the HIV Latency and the Role of Cytokines. Pathogens 2019, 8, 137.
- Hokello, J.; Sharma, A.L.; Tyagi, M. Combinatorial Use of Both Epigenetic and Non-Epigenetic Mechanisms to Efficiently Reactivate HIV Latency. Int. J. Mol. Sci. 2021, 22, 3697.
- North, T.W.; Higgins, J.; Deere, J.D.; Hayes, T.L.; Villalobos, A.; Adamson, L.; Shacklett, B.L.; Schinazi, R.F.; Luciw, P.A. Viral sanctuaries during highly active antiretroviral therapy in a nonhuman primate model for AIDS. J. Virol. 2010, 84, 2913–2922.
- Spudich, S.; Gisslen, M.; Hagberg, L.; Lee, E.; Liegler, T.; Brew, B.; Fuchs, D.; Tambussi, G.; Cinque, P.; Hecht, F.M.; et al. Central nervous system immune activation characterizes primary human immunodeficiency virus 1 infection even in participants with minimal cerebrospinal fluid viral burden. J. Infect. Dis. 2011, 204, 753–760.
- Lustig, G.; Cele, S.; Karim, F.; Derache, A.; Ngoepe, A.; Khan, K.; Gosnell, B.I.; Moosa, M.S.; Ntshuba, N.; Marais, S.; et al. T cell derived HIV-1 is present in the CSF in the face of suppressive antiretroviral therapy. PLoS Pathog. 2021, 17, e1009871.
- Peluso, M.J.; Ferretti, F.; Peterson, J.; Lee, E.; Fuchs, D.; Boschini, A.; Gisslen, M.; Angoff, N.; Price, R.W.; Cinque, P.; et al. Cerebrospinal fluid HIV escape associated with progressive neurologic dysfunction in patients on antiretroviral therapy with well controlled plasma viral load. AIDS 2012, 26, 1765–1774.
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362.
- Ko, A.; Kang, G.; Hattler, J.B.; Galadima, H.I.; Zhang, J.; Li, Q.; Kim, W.K. Macrophages but not Astrocytes Harbor HIV DNA in the Brains of HIV-1-Infected Aviremic Individuals on Suppressive Antiretroviral Therapy. J. Neuroimmune Pharmacol. 2019, 14, 110–119.
- Reu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784.
- Veenstra, M.; Leon-Rivera, R.; Li, M.; Gama, L.; Clements, J.E.; Berman, J.W. Mechanisms of CNS Viral Seeding by HIV+ CD14+ CD16+ Monocytes: Establishment and Reseeding of Viral Reservoirs Contributing to HIV-Associated Neurocognitive Disorders. mBio 2017, 8, e01280-17.
- Leon-Rivera, R.; Veenstra, M.; Donoso, M.; Tell, E.; Eugenin, E.A.; Morgello, S.; Berman, J.W. Central Nervous System (CNS) Viral Seeding by Mature Monocytes and Potential Therapies To Reduce CNS Viral Reservoirs in the cART Era. mBio 2021, 12, e03633-20.
- Kruize, Z.; Kootstra, N.A. The Role of Macrophages in HIV-1 Persistence and Pathogenesis. Front. Microbiol. 2019, 10, 2828.
- Whitney, J.B.; Hill, A.L.; Sanisetty, S.; Penaloza-MacMaster, P.; Liu, J.; Shetty, M.; Parenteau, L.; Cabral, C.; Shields, J.; Blackmore, S.; et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 2014, 512, 74–77.
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 latency in monocytes/macrophages. Viruses 2014, 6, 1837–1860.
- Alvarez-Carbonell, D.; Ye, F.; Ramanath, N.; Garcia-Mesa, Y.; Knapp, P.E.; Hauser, K.F.; Karn, J. Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog. 2019, 15, e1008249.
- Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14, 9.

