1. Introduction
Human immunodeficiency virus type-1 (HIV-1) remains one of the most serious public health challenges [1,2,3,4]. HIV-1 invades the brain soon following systemic infection. Several mechanisms have been suggested for HIV-1 entry into the central nervous system (CNS). However, the “Trojan horse hypothesis”, which states that HIV-1 infection of the brain occurs through migration of the infected cells across the blood–brain barrier (BBB), is the most favored. Although CD4+ T cells, as well as monocytes and macrophages, are the primary cellular targets for productive HIV-1 infection [5,6,7], in the brain, macrophages and microglia are the main cell types productively infected by HIV-1, and virus production in the CNS is not seen before the onset of acquired immunodeficiency syndrome (AIDS) [8,9].
2. HIV Transcriptional Regulation in the CNS
The HIV-1 long terminal repeat (LTR) is responsible for HIV transcriptional initiation, and it contains
cis-regulatory elements that specifically bind the transcription factors involved in the regulation of HIV transcription. The LTR is comprised of three distinct regions, including the unique 3′ (U3) end, repeated (R) sequence, and the unique 5′ (U5) end. The U3 is made up of elements that mediate RNA polymerase II (RNAP II) binding to the viral template DNA and the TATA box located at the −28 nucleotide position relative to the transcription start site. Two nuclear factor kappa beta (NF-κB) and three specificity protein 1 (Sp1)-binding sites are found at the 5′ end of the TATA box [
4,
13,
14]. The HIV transcription is initiated from the LTR following the binding of a highly conserved 38-KD TATA box binding protein (TBP) to the TATA box sequence. TBP binding mediates the recruitment of additional transcription factors forming the preinitiation complex referred to as the TBP-associated factor (TAF). The TBP and TAF form a multiprotein complex referred to as transcription factor II D (TFIID), which is the minimal protein complex capable of inducing basal transcription from the HIV LTR.
It is, however, worth noting that the efficient initiation of transcription from the HIV LTR requires the interaction of TFIID with the upstream enhancer-binding transcription factors, such as NF-κB or the nuclear factor of activated T cells (NFAT) [
14,
15]. Transcription factor II H (TFIIH) then phosphorylates the C-terminal domain (CTD) of RNAP II within the transcription initiation complex in order to enhance HIV transcriptional elongation [
14,
16,
17]. However, when the viral transcription trans-activator protein Tat is absent, HIV transcription is less efficient, because RNAP II stalls just a few nucleotides from the transcription start site following the promoter clearance. In the absence of the viral Tat protein, transcription initiation from the HIV LTR is efficient; however, the transcription is impaired, because the promoter engages poorly processive RNAP II, which disengages prematurely from the DNA template [
18]. On the other hand, when Tat is present, the synthesis of viral messenger RNA (mRNA) is enhanced because of an increased elongation efficiency mediated by Tat [
18,
19,
20,
21]. In what is referred to as TAR-dependent HIV transcription, Tat acts by binding to an RNA element known as the transcription response (TAR) element, which is a stem loop structure that forms at the 5′ end of nascent viral RNA transcripts following transcription through the first 59 nucleotides [
14]. Tat binds to TAR and recruits positive transcription elongation factor b (P-TEFb), a complex comprising cyclin T1 and cyclin-dependent kinase-9 (CDK9); the kinase subunit then hyperphosphorylates the CTD of the largest subunit of RNAP II, resulting in enhanced RNAP II processivity and transcription efficiency [
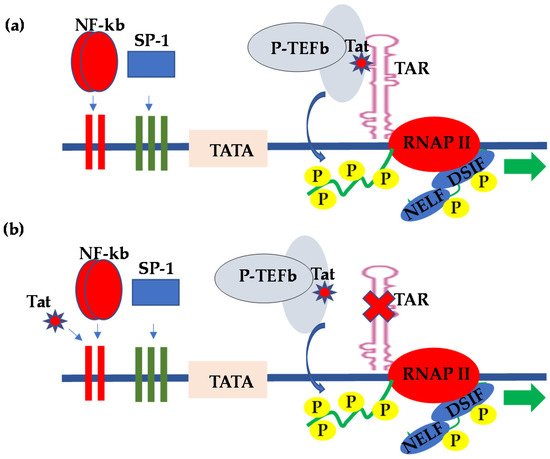
15] (
Figure 1a). In order to provide more insight into HIV pro-viral transcriptional regulation, Schulze-Gahmen and Hurley [
22] determined the crystal structure of the super elongation complex in a complex with HIV Tat and TAR RNA. They observed that, in the crystal structure, the role of Tat is three-fold—namely, scaffolding and stabilization of the Tat-TAR recognition motif (TRM), making specific interactions through its zinc-coordinating loop and making electrostatic interactions through its arginine-rich motif (ARM). Interestingly, most recently, Hokello et al. [
23] demonstrated that the cellular transcription factor, activator protein-1 (AP-1), synergizes with NF-κB to regulate HIV transcriptional elongation following T-cell receptor activation.
Figure 1. Molecular model for the (a) TAR-dependent and (b) TAR-independent regulation of HIV transcription.
The HIV Tat provided the first example of viral gene expression regulation through the control of elongation by RNAP II. Although HIV Tat stimulates HIV transcriptional elongation primarily through specific interactions with TAR, and HIV-1 transcriptional regulation by Tat in most cell types requires intact TAR sequences [
18], reports from several groups subsequently demonstrated that Tat regulation of HIV LTR-derived transcription in CNS-derived astrocytes and glial cells occurs in the absence of TAR, a mechanism of HIV transcriptional regulation referred to as TAR-independent transcription [
24,
25]. In this regard, numerous reports have subsequently demonstrated that the upstream transcription elements within the LTR were responsible for the Tat-mediated activation of HIV LTR transcription in the absence of the TAR element (
Figure 1b). Specifically, the CNS-derived cells were found to exhibit kappa B-binding activity, which interacted with Tat to activate LTR transcription [
24,
26,
27,
28]. Indeed, the kappa B binding transcription factor from the CNS cells consisted of components that were indistinguishable from the prototypical NF-κB [
26]. The TAR-independent Tat regulation of HIV transcriptional activity mediated by the kappa B-binding site provided implications with respect to the ability of Tat to alter cellular gene expressions and possibly contribute to a myriad of conditions associated with HIV infection, including the altered immune status, CNS toxicity, and tumor formation. Consistent with the above observations, other reports demonstrated that the HIV-1 Tat protein released from HIV-infected cells could be taken up by uninfected cells only to exert its effects on the responsive genes. For instance, Cupp et al. [
29] and Sawaya et al. [
30] demonstrated that HIV-1 Tat regulates the expression of transforming growth factor beta-1 (TGF-β1) in human astrocytic glial cells. TGF-β1 is a cytokine with potent immunosuppressive activity.
Gray et al. [
31] also reported that the CNS-derived HIV-1 strains exhibit polymorphisms within the Sp1-binding sequence within the HIV LTR. These mutations resulted in decreased binding of the Sp1 transcription factor to the CNS-derived LTRs, thus reducing the transcriptional response of CNS-derived viruses. Accordingly, this observation suggests that the Sp1 transcription factor, which is critically important in HIV transcriptional regulation in many cell types, may not be required in HIV transcriptional regulation in cells derived from the CNS.
On the other hand, drugs of abuse, including cocaine, amphetamine, methamphetamine, heroin, and morphine, have been reported to upregulate HIV transcription, particularly in the CNS, and that illicit drug use correlates well with the high rate of HIV transmission among illicit drug users, as well as the rapid rate of acquired immunodeficiency syndrome (AIDS) progression [
32,
33,
34,
35]. Sahu et al. [
36] demonstrated that cocaine, for instance, promotes both the initiation and elongation phases of HIV-1 transcription by activating NF-κB and mitogen and stress-activated kinase-1 (MSK-1). MSK-1 is responsible for the phosphorylation of the histone H3 and the p65 subunit of NF-κB, which subsequently enhances NF-κB interactions with p300, thus promoting the recruitment of P-TEFb. P-TEFb is a cellular cofactor for the viral Tat protein, which mediates HIV transcriptional elongation. In addition to the above elaborate mechanism of how drugs of abuse enhance HIV transcription, the observation that illicit drugs activate NF-κB is consistent with the fact that NF-κB mediates the TAR-independent Tat regulation of HIV transcription in cells derived from the CNS. Given these observations, it is clearly evident that HIV transcriptional regulation in the CNS is, in many ways, different from that of many cell types. This is especially so because different tissues are made up of specialized cells that are formed through cellular differentiation. As such, different types of cells contain different types of transcription factors, and this is the reason why there may be differential regulations of HIV transcription in the different anatomical sites represented in
Table 1.
Table 1. The different anatomical sites that harbor latent HIV provirus reservoirs.
| Anatomic Sites |
Specific Sites |
| Brain |
|
| Primary Lymphatic tissue/organ |
Thymus and Bone marrow |
| Non-Lymphoid tissues |
Liver, Kidney, Adipose, reproductive tract and other |
| Secondary lymphatic tissue/organs |
Tonsils, Adenoids, spleen, Mucosa-associated lymphatic tissues and lymph nodes |
| Peripheral Blood |
|
3. HIV Latency in the CNS
The latent HIV provirus, the hallmark of HIV latency, is integrated viral DNA that is transcriptionally silent and cannot be reached by the current combination antiretroviral therapy (cART), thus posing a great obstacle to the treatment and cure of HIV infection [
37,
38,
39,
40,
41,
42,
43,
44]. Attempts to cure HIV infection in patients undergoing cART require the effective targeting of all possible viral reservoirs in all anatomical sites. Other than the memory CD4+ T cells, several HIV reservoirs have been identified in different anatomical sites (
Table 1) where HIV-1 continues to replicate despite cART. For instance, using the nonhuman primate model infected with RT-SHIV, a chimera of simian immunodeficiency virus containing the HIV-1 reverse transcriptase sequences, North et al. [
45] demonstrated that HIV DNA and viral RNA were present in RT-SHIV-infected macaques that were treated with potent cART regimens consisting of efavirenz, emtricitabine, and tenofovir. Moreover, an additional analysis provided evidence for the presence of full-length viral RNA in tissues of the animals with the virus suppressed by cART. Interestingly, they showed that the highest levels of HIV DNA and viral RNA in cART-treated macaques were in lymphoid tissues—particularly the spleen, lymph nodes, and gastrointestinal tract—demonstrating that there is widespread persistence, as well as residual viral replication, in different anatomical sites, even during cART. In particular, the CNS is compartmentalized and serves as a specific site of HIV infection. Understanding of the cellular populations that harbor latent HIV infection in the CNS is critically important in attempts to find solutions to HIV latency.
HIV-1 infection of the CNS has peculiar characteristics in that CNS infection by HIV-1 can be significant and occurs very early following viral transmission. Secondly, local HIV replication in the CNS can be quite diverse and evolves over time. Thirdly, HIV-1 may persist in the CNS, even in the face of cART, due to insufficient penetration of the CNS by the current cART drugs but, also, due to the long-lived nature of the resident CNS cells. The physiological effects of HIV infection in the CNS are seen in patients with HIV-associated neurocognitive disorders (HAND) and HIV-associated dementia (HAD), which have been linked to increased neurological injury due to inflammation.
Early, following infection, HIV-1 can be found in the cerebrospinal fluid (CSF) at very low levels with very minimal viral burden in the CNS [
46]. Therefore, this suggests that HIV most likely enters the CNS/CSF at low levels through the incomplete partitioning of the virus at the BBB or through background trafficking of the immune cells, including infected CD4+ T cells. Although the CNS is a site where HIV persists despite cART, persistence can also be maintained in the form of CSF escape, a scenario whereby HIV is detectable in the CSF but undetectable in the blood in some patients. For instance, Lustig et al. [
47] reported a high prevalence of CSF escape of 28% in cART-treated HIV patients in South Africa. During another development, Peluso et al. [
48] observed that the development of neurologic symptoms in HIV patients on cART with low or undetectable plasma viremia was an indication of CSF escape. The CSF, therefore, serves as the reservoir of the virus that replenishes the CNS or the blood.
Wallet et al. [
49] observed that microglial cells, which are the resident macrophages in the CNS, are one of the major cellular reservoirs of HIV latency. Similarly, Ko et al. [
50] demonstrated that it is the microglial cells that harbor HIV DNA in the brains of HIV-1-infected aviremic individuals on suppressive cART. Microglial cells are also the most abundant mononuclear macrophages found in the brain parenchyma. The median renewal rate of these cells is about 30% per year, and most of the cells in this population regenerate throughout a lifetime [
51]. It is believed that microglial cells are infected by HIV-1 through the transmigration of infected monocytes, which occur very early in the course of HIV infection. Indeed, recent reports have identified a specific subset of HIV-infected monocytes that were HIV+, CD14+, and CD16+ that preferentially crossed the BBB [
52]. Similarly, Leon-Rivera et al. [
53] most recently characterized the mechanism of CNS viral reservoir establishment and replenishment using the peripheral blood mononuclear cells (PBMCs) of HIV patients on prolonged cART and established that monocytes from PBMCs with integrated HIV proviruses that were transcriptionally active selectively transmigrated across the human BBB model. In the CNS, replication-competent HIV-1 is reported to predominantly persist in resident macrophages, including microglia and perivascular monocytes/macrophages, whereby they disseminate the virus to other cell types. For a detailed review on the role of macrophages in the persistence of HIV pathogenesis, please refer to Kruize and Kootstra [
54]. The infection and establishment of HIV latency in microglial cells seem to occur very early in the evolution of HIV infection [
55]. The features of microglial cells, which allow for HIV persistence in the brain, include a resistance to cytopathic effects and the fact that they are nonlytic and resistant to apoptosis induced by HIV infection [
56].
Using a cellular coculture system, the Karn group investigated the hypothesis that HAND resulted from the periodic emergence of HIV from a latency within microglial cells in response to neuronal damage or inflammatory signals. In this regard, when clonal human microglial cells (HC69) infected with HIV (hµglia/HIV) were cocultured for a short time with healthy neurons, HIV was silenced. However, the neuron-dependent induction of HIV latency in HC69 cells was demonstrated using induced pluripotent stem cell-derived GABAergic cortical and dopaminergic, but not motor, neurons, whereby damaged neurons induced HIV expression in latently infected microglial cells. Following 48–72 h of a coculture, low levels of HIV gene expression further damaged the neurons with a result that further enhanced HIV expression [
57]. In a separate set of experiments, Alvarez-Carbonelli et al. [
58] demonstrated that Toll-like receptor (TLR) signaling—in particular, TLR-3 activation—can efficiently reactivate HIV transcription in infected microglia but not in monocytes or T cells.
This entry is adapted from the peer-reviewed paper 10.3390/vaccines9111272