Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yukihito Higashi | -- | 1981 | 2023-02-02 11:07:53 | | | |
| 2 | Rita Xu | Meta information modification | 1981 | 2023-02-03 03:03:46 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Higashi, Y. Vascular Endothelial Dysfunction-Related Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/40768 (accessed on 29 July 2026).
Higashi Y. Vascular Endothelial Dysfunction-Related Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/40768. Accessed July 29, 2026.
Higashi, Yukihito. "Vascular Endothelial Dysfunction-Related Disease" Encyclopedia, https://encyclopedia.pub/entry/40768 (accessed July 29, 2026).
Higashi, Y. (2023, February 02). Vascular Endothelial Dysfunction-Related Disease. In Encyclopedia. https://encyclopedia.pub/entry/40768
Higashi, Yukihito. "Vascular Endothelial Dysfunction-Related Disease." Encyclopedia. Web. 02 February, 2023.
Copy Citation
Oxidative stress and chronic inflammation play an important role in the pathogenesis of atherosclerosis. Atherosclerosis develops as the first step of vascular endothelial dysfunction induced by complex molecular mechanisms. Vascular endothelial dysfunction leads to oxidative stress and inflammation of vessel walls, which in turn enhances vascular endothelial dysfunction. Vascular endothelial dysfunction and vascular wall oxidative stress and chronic inflammation make a vicious cycle that leads to the development of atherosclerosis.
oxidative stress
inflammation
endothelial function
1. Introduction
Oxidative stress and inflammation play an important role in the development of cardiovascular diseases (CVD) [1][2][3][4][5]. The sources of production of reactive oxygen species (ROS) in blood vessels include nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase, uncoupled endothelial nitric oxide synthase (eNOS), arachidonic acid metabolic pathway, mitochondrial electron transfer system dysfunction, and catecholamine autoxidation [6][7]. Among these, NADPH oxidase is the most important and is involved in the production of ROS in vivo, and it is activated by various cytokines, vasoactive substances, and shear stress [8][9]. It is well known that ROS have opposing effects on cell viability. ROS induce cell proliferation, hypertrophy, migration, and apoptosis [10][11]. Chronic inflammation, which begins as a biological response to vascular endothelial dysfunction, is also thought to be the primary cause of atherosclerosis [3][4][5]. Various factors including oxidative stress, oxidized low-density lipoprotein (LDL), thrombi, and viral and bacterial infections induce acute and chronic inflammatory cell infiltrates, including neutrophils, lymphocytes, and macrophages, which in turn enhance local vascular inflammation by enhancing the production of inflammatory cytokines by the infiltrating inflammatory cells [12][13]. It is thought that in the presence of CVD, chronic inflammation makes a vicious cycle, leading to the maintenance and progression of atherosclerosis. Therefore, evaluation of vascular function, which is the primary point of action of chronic inflammation, is extremely important for elucidating the pathogenesis of CVD, determining therapeutic efficacy, and predicting prognosis.
2. Vascular Endothelial Function
The vascular endothelium is anatomically located in the innermost layer of blood vessels, and the vascular system, a closed system consisting of the heart, arteries, and veins, is composed entirely of one cell layer. Endothelium cells secrete vasodilators including nitric oxide (NO), prostaglandin I2, C-type natriuretic peptide, and endothelium-derived vascular hyperpolarizing factor as well as vasoconstrictors including endothelin, angiotensin II, prostaglandin H2, and thromboxane A2 [14][15]. Among these bioactive substances, NO plays a very important role in atherosclerosis. NO is produced and secreted from L-arginine, an essential amino acid, by activation of endothelial NO synthase (eNOS) through mechanical stimulation of vascular endothelial cells or binding to receptors by agonists such as acetylcholine, histamine, and bradykinin or by shear stress from blood flow in vascular endothelial cells [16][17]. The secreted gaseous NO is transmitted by diffusion to nearby VSMCs and activates intracellular soluble guanylate cyclase, which increases the amount of cyclic guanosine monophosphate and relaxes vascular smooth muscle [18]. The normal vascular endothelium maintains vascular function and structure by adjusting the balance between vasodilation and vasoconstriction, proliferation and antiproliferation of VSMCs, coagulation and anticoagulation, inflammation and anti-inflammation, and oxidation and antioxidation [19]. If all of the vascular endothelium in the body could be collected, the total weight would be equivalent to that of the liver. If it could be spread over an entire surface, the total area would be equivalent to six tennis courts and if it could be connected in a row, the length would be 100,000 km or two and a half times around the earth [20]. Atherosclerosis develops and progresses the first stage of vascular endothelial dysfunction [20]. Hypertension, diabetes, dyslipidemia, aging, obesity, smoking, lack of exercise, excessive salt intake, and menopause induce vascular endothelial dysfunction [21]. Aging is the largest factor that determines vascular endothelial function [22]. Vascular endothelial function is now recognized as a predictive factor in the development of CVD. A meta-analysis by Lerman et al. [23] confirmed that endothelial function is an independent predictor of cardiovascular complications. Vascular endothelial function can also be viewed as a target for treatment of atherosclerosis. Endothelial dysfunction is not irreversible and can be corrected with appropriate interventions such as antihypertensive medications, antidiabetic agents, lipid-lowering therapy, and lifestyle modification [24][25][26][27][28][29][30]. Improvement in endothelial dysfunction is expected to reduce the incidence of coronary and cerebrovascular events and improve life expectancy in the future. Figure 1 shows the roles of oxidative stress and inflammation in the process of endothelial dysfunction-related disease.
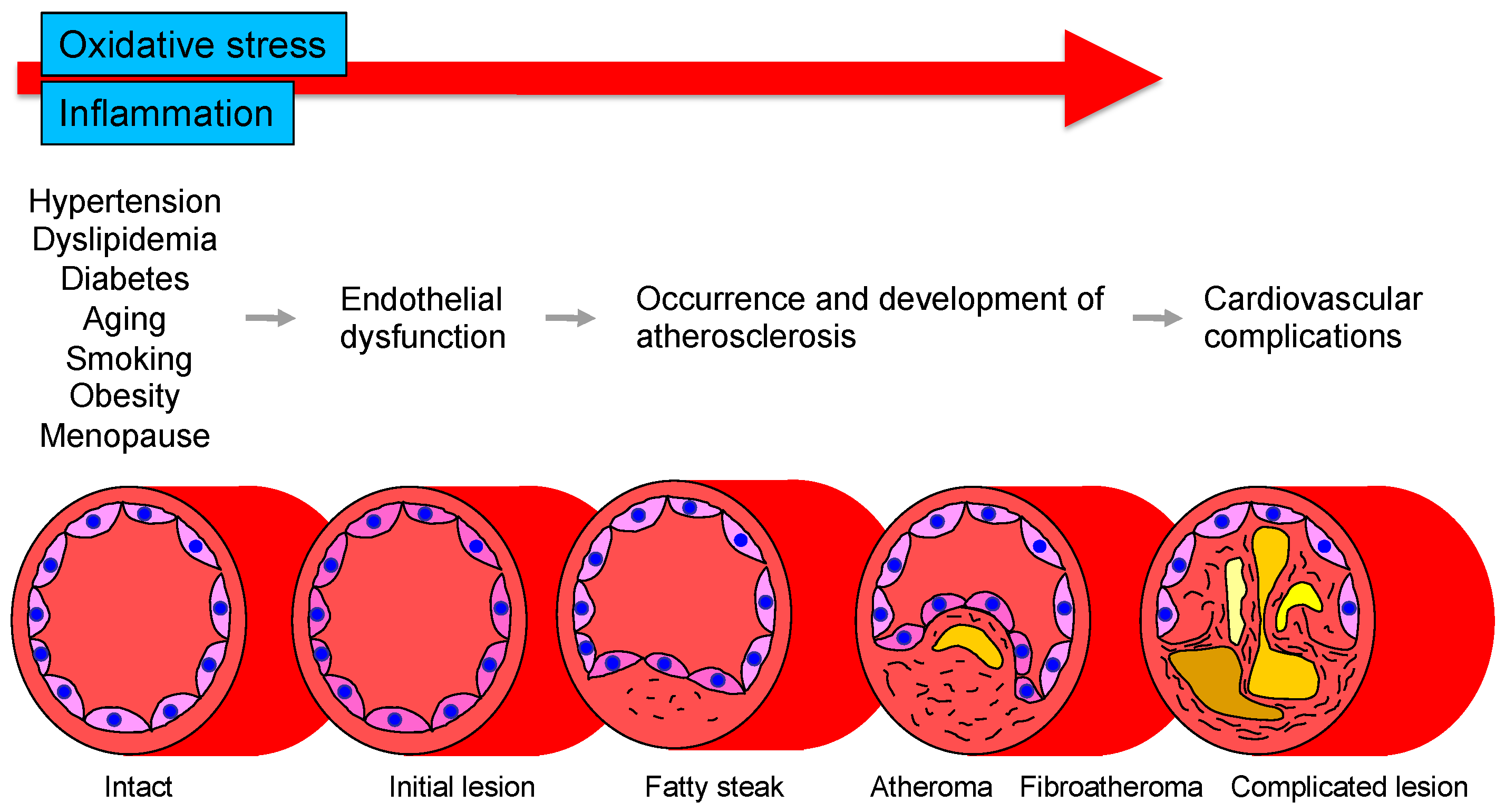
Figure 1. Roles of oxidative stress and inflammation in the process of endothelial dysfunction-related disease.
Several methods for measuring vascular endothelial function are currently used [31][32]. These can be broadly classified into methods that evaluate blood flow and vessel diameter by reactive hyperemia after ischemia and methods that evaluate reactivity by administering a bioactive substance. Unfortunately, there is no gold standard method, but at this stage, plethysmography is considered to best reflect vascular endothelial function. In general, this method evaluates vascular endothelial function by measuring changes in blood flow through selective administration of NO-producing stimulants such as acetylcholine, methacholine, bradykinin, histamine, or NO inhibitors to arteries in the limbs. Various vasoactive substances can be used in this method in addition to NO agonists and antagonists, allowing multifaceted evaluation of the dynamics of various bioactive substances produced and secreted by vascular endothelial cells, rather than examining only NO from a single perspective. The ultrasound-based method, flow-mediated vasodilation (FMD), is evaluated by the change in vessel diameter after reactive hyperemia in an extremity. FMD (% change) is calculated by [(maximum vessel diameter after lifting of the frame − baseline vessel diameter)/baseline vessel diameter] × 100. Measurement of peripheral arterial pulse amplitude in fingers is also used to assess endothelial function. The ratio of peripheral arterial pulse amplitude before and after reactive hyperemia (reactive hyperemia index) is calculated. Measurement of biomarkers in blood or urine is the most convenient and non-invasive method, but, unfortunately, there are currently no biomarkers that can be evaluated. However, there are various problems such as the possibility that they do not directly reflect NO production and the accuracy of measurement. These measurements should be considered as adjuncts to endothelial function assessment using FMD or plethysmography. FMD measurement is currently the most widely used method for evaluating vascular endothelial function and is likely to become more widely used in the future. However, it is also true that the method still has many issues to be resolved and many problems to be solved in the future, including the issue of reproducibility. Each method has its own advantages and disadvantages, and it is desirable to improve, refine, and standardize the method or to develop a new concept of a measurement device [32].
3. Oxidative Stress
3.1. Oxidative Stress and Vascular Injury
Oxidative stress plays an important role in CVD and CV events through various mechanisms including activation of NADPH oxidase, xanthine oxidase and cyclooxygenase, dysfunction of the mitochondrial electron transfer system, uncoupled eNOS, catecholamine autoxidation and failure of the antioxidative system (Figure 2). Circulating LDL enters the subendothelium and undergoes denaturation by oxidative stress to become oxidized LDL [12][13]. Oxidized LDL induces the expression of monocyte migration factor macrophage chemotactic protein 1 (MCP-1) in vascular endothelial cells and also induces the expression of adhesion factors such as intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), causing peripheral monocytes to adhere to the endothelium and invade the subendothelium [33]. Oxidized LDL also induces the secretion and production of macrophage colony-stimulating factor by vascular endothelial cells, leading to the maturation and differentiation of monocytes into macrophages. Oxidized LDL induces apoptosis in many cells, but it has been observed that a large amount of oxidized LDL accumulates in macrophages, leading to foam cell formation without inducing apoptosis [34]. It is thought that these foam cells release various cytokines, inducing inflammation and oxidative stress in the vascular endothelium. In atherosclerotic lesions, these systems make a vicious cycle that leads to the formation of atheroma and, in the final stage, to CV events. Oxidized LDL has also been reported to promote apoptosis of VSMCs lining the luminal side of the atheroma (thinning of the fibrous membrane), which may contribute to atheroma fragility and instability [35]. Lysophosphatidylcholine produced during oxidative degeneration of LDL impairs the NO synthesis system and contributes to the decrease in NO production [36]. Oxidized LDL is also known to act in thrombus formation by increasing plasminogen activator inhibitor-1 (PAI-1) production and decreasing tissue plasminogen activator (t-PA) production from vascular endothelial cells [37]. Thus, oxidized LDL is widely involved in the initiation, maintenance, and progression of atherosclerosis and atherogenesis. Lectin-like oxidized LDL receptor-1 (LOX-1), a novel scavenger receptor for oxidized LDL, was cloned in vascular endothelial cells [38]. In vitro, LOX-1 expression is known to be induced by various cytokines and shear stress [39]. Interestingly, LOX-1 is rarely expressed in normal vessels but is strongly expressed in endothelial cells, VSMCs and macrophages in atherosclerotic lesions [39][40].
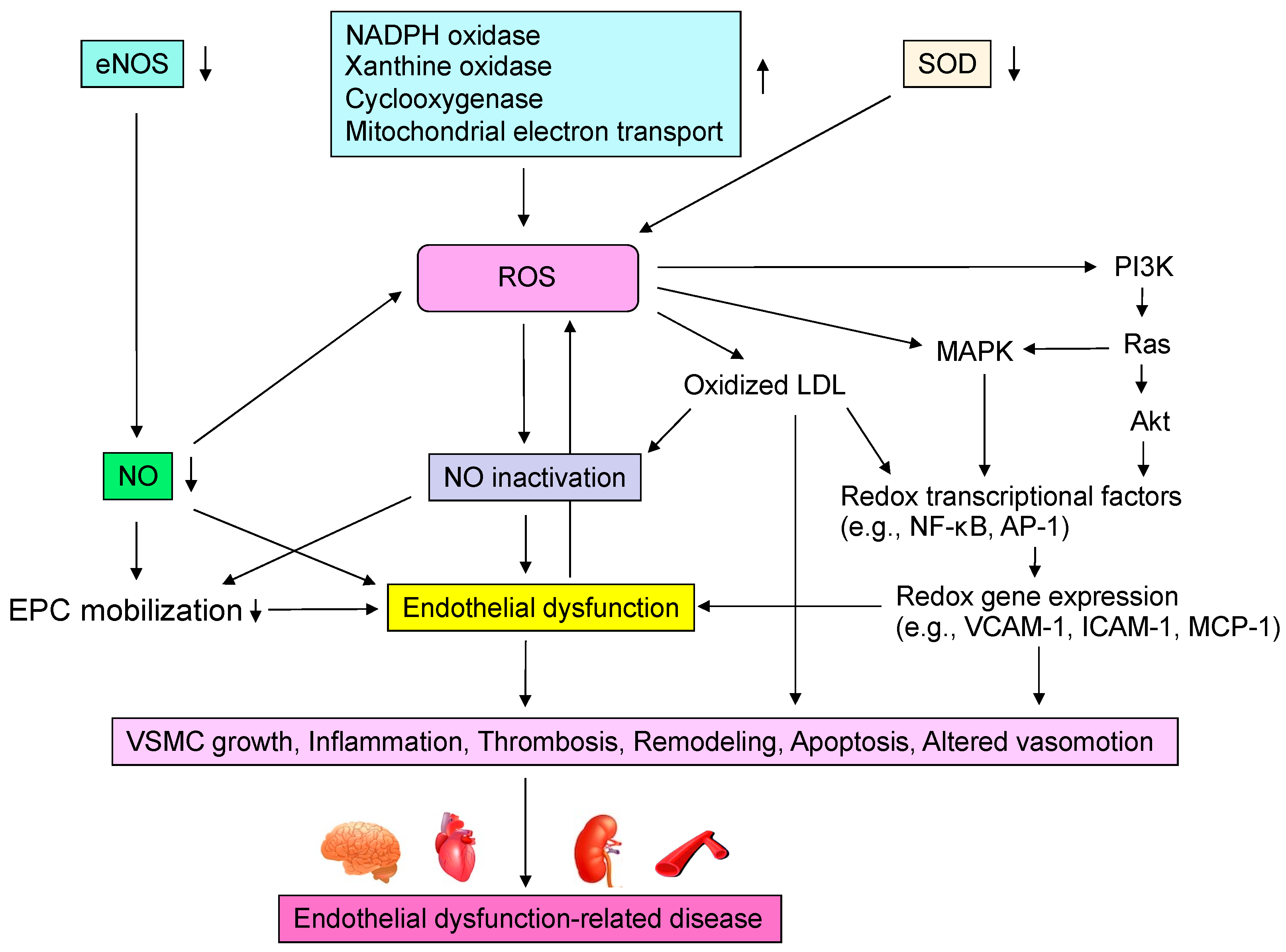
Figure 2. Mechanisms by which reactive oxygen species (ROS) induce endothelial dysfunction-related disease. eNOS, endothelial nitric oxide synthase; NO, nitric oxide; NADPH, nicotinamide adenine dinucleotide phosphate; SOD, superoxide dismutase; LDL, low-density lipoprotein; MAPK, mitogen-activated protein kinase kinases; NF-κB, nuclear factor-kappa B; AP-1, activator protein-1; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intracellular adhesion molecule-1; MCP-1, macrophage chemotactic protein 1; EPC, endothelial progenitor cell; VSMC, vascular smooth muscle cell.
3.2. ROS Metabolism and Oxidative Stress
ROS are composed of free radicals such as superoxide anions, hydroxyl radicals, and NO and free radical metabolites such as hydrogen peroxide and peroxynitrite. Free radicals have one or more unpaired electrons and are much more reactive than non-radicals. ROS are usually kept at low concentrations as signaling molecules, but when produced in excess, they can cause a variety of adverse effects. In a healthy cellular environment, the toxicity associated with excess production of ROS is eliminated by the operation of antioxidant systems such as superoxide dismutase, catalase and glutathione peroxidase [41]. Failure of the antioxidant defense system or excessive free-radical production that exceeds the antioxidants can lead to cellular damage, and administration of antioxidants can be protective against cellular damage [42]. Oxidative stress is defined as a state in which the balance between oxidation and anti-oxidation is upset by a failure of the antioxidant defense system or by an excess of ROS production that exceeds that of antioxidants, and the balance is tilted toward a relative excess of ROS. ROS are diverse in terms of their production systems, operability, and chemical reactivity. When two free radicals react, they share an unpaired electron with each other to form free radical metabolites. In addition, radicals and free radical metabolites initiate chain reactions to form new radicals.
3.3. Enzymes Related to the Production of ROS
NADPH oxidase, also known as the Nox enzyme group, is the most important enzyme in the vascular ROS-producing system (Figure 2) [43]. In the inactivated state, p47phox and p67phox are located in the cytoplasm, while p22phox and Nox2 are located at the plasma membrane [43]. Upon mechanical stimuli such as angiotensin II, endothelin-1, thrombin, or pressure loading, p47phox is phosphorylated and p67phox is activated. Phox is phosphorylated, and the cytoplasmic component complex is translocated into the cytoplasm, where it assembles its components to generate enzymatic activity as NADPH oxidase [44]. This requires the guanine nucleotide-binding proteins Rac-2 and Rap-1 [43]. Activation of NADPH oxidase results in the production of superoxide from oxygen. Usio-Fukai et al. [45] demonstrated that p22phox is involved in NADPH oxidase-mediated production of ROS in the aorta of hypertensive rats and that it regulates angiotensin II-induced vascular smooth muscle thickening. Recently, Touyz et al. [46] reported that VSMCs obtained from six human resistance vessels expressed Nox2 upon angiotensin II stimulation. Nox1, a homologue of Nox2, is expressed in human aortic VSMCs and Wistar-Kyoto rat celiac artery smooth muscle cells but not in human resistance vessel smooth muscle cells [47]. Nox4, another homologue of Nox2, is present in both cell types. Furthermore, other NADPH oxidase subunits, including p40phox, p47phox, p67phox and p22phox, have been found to be present in human resistance artery VSMCs [47]. NADPH oxidase is a novel enzyme that is involved in the production of ROS in human vascular endothelial cells and VSMCs [48][49]. NADPH oxidase is still unknown, and researchers are waiting for more information to be accumulated.
References
- Meigs, J.B.; Larson, M.G.; Fox, C.S.; Keaney, J.F., Jr.; Vasan, R.S.; Benjamin, E.J. Association of oxidative stress, insulin resistance, and diabetes risk phenotypes: The Framingham Offspring Study. Diabetes Care 2007, 30, 2529–2535.
- Murthy, V.L.; Yu, B.; Wang, W.; Zhang, X.; Alkis, T.; Pico, A.R.; Yeri, A.; Bhupathiraju, S.N.; Bressler, J.; Ballantyne, C.M.; et al. Molecular Signature of Multisystem Cardiometabolic Stress and Its Association with Prognosis. JAMA Cardiol. 2020, 5, 1144–1153.
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762.
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621.
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979.
- Li, H.; Horke, S.; Förstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219.
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kröller-Schön, S.; Steven, S.; Schulz, E.; Münzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689.
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal 2009, 11, 791–810.
- Rochfort, K.D.; Collins, L.E.; McLoughlin, A.; Cummins, P.M. Shear-dependent attenuation of cellular ROS levels can suppress proinflammatory cytokine injury to human brain microvascular endothelial barrier properties. J. Cereb. Blood Flow Metab. 2015, 35, 1648–1656.
- Li, J.M.; Shah, A.M. Endothelial cell superoxide generation: Regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1014–R1030.
- García-Redondo, A.B.; Aguado, A.; Briones, A.M.; Salaices, M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacol. Res. 2016, 114, 110–120.
- Lubrano, V.; Balzan, S. Roles of LOX-1 in microvascular dysfunction. Microvasc. Res. 2016, 105, 132–140.
- Hwang, J.; Ing, M.H.; Salazar, A.; Lassègue, B.; Griendling, K.; Navab, M.; Sevanian, A.; Hsiai, T.K. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: Implication for native LDL oxidation. Circ. Res. 2003, 93, 1225–1232.
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376.
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269.
- Lüscher, T.F. Imbalance of endothelium-derived relaxing and contracting factors. Am. J. Hypertens. 1990, 3, 317–330.
- Vane, J.R.; Anggard, E.E.; Botting, R.M. Regulatory functions of the vascular endothelium. N. Engl. J. Med. 1990, 323, 27–36.
- Golshiri, K.; Ataei Ataabadi, E.; Portilla Fernandez, E.C.; Jan Danser, A.H.; Roks, A.J.M. The importance of the nitric oxide-cGMP pathway in age-related cardiovascular disease: Focus on phosphodiesterase-1 and soluble guanylate cyclase. Basic Clin. Pharmacol. Toxicol. 2020, 127, 67–80.
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126.
- Higashi, Y.; Noma, K.; Yoshizumi, M.; Kihara, Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ. J. Off. J. Jpn. Circ. Soc. 2009, 73, 411–418.
- Higashi, Y.; Yoshizumi, M. Exercise and endothelial function: Role of endothelium-derived nitric oxide and oxidative stress in healthy subjects and hypertensive patients. Pharmacol. Ther. 2004, 102, 87–96.
- Benjamin, E.J.; Larson, M.G.; Keyes, M.J.; Mitchell, G.F.; Vasan, R.S.; Keaney, J.F., Jr.; Lehman, B.T.; Fan, S.; Osypiuk, E.; Vita, J.A. Clinical correlates and heritability of flow-mediated dilation in the community: The Framingham Heart Study. Circulation 2004, 109, 613–619.
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368.
- De Filippis, E.; Cusi, K.; Ocampo, G.; Berria, R.; Buck, S.; Consoli, A.; Mandarino, L.J. Exercise-induced improvement in vasodilatory function accompanies increased insulin sensitivity in obesity and type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2006, 91, 4903–4910.
- Sanada, M.; Higashi, Y.; Nakagawa, K.; Sasaki, S.; Kodama, I.; Sakashita, T.; Tsuda, M.; Ohama, K. Hormone replacement effects on endothelial function measured in the forearm resistance artery in normocholesterolemic and hypercholesterolemic postmenopausal women. J. Clin. Endocrinol. Metab. 2002, 87, 4634–4641.
- Kimura, M.; Goto, C.; Umemura, T.; Ueda, K.; Nishioka, K.; Jitsuiki, D.; Yoshizumi, M.; Chayama, K.; Higashi, Y. Repetition of ischemic preconditioning augments endothelium-dependent vasodilation in humans: Role of endothelium-derived nitric oxide and endothelial progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1403–1410.
- Fujimura, N.; Jitsuiki, D.; Goto, C.; Soga, J.; Hidaka, T.; Umemura, T.; Nishioka, K.; Oshima, T.; Chayama, K.; Higashi, Y. Geranylgeranylacetone, Hsp90/AMPK/eNOS/NO pathway, and endothelial function in humans. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 153–160.
- Hambrecht, R.; Wolf, A.; Gielen, S.; Linke, A.; Hofer, J.; Erbs, S.; Schoene, N.; Schuler, G. Effect of exercise on coronary endothelial function in patients with coronary artery disease. N. Engl. J. Med. 2000, 342, 454–460.
- Goto, C.; Nishioka, K.; Umemura, T.; Jitsuiki, D.; Sakaguchi, A.; Kawamura, M.; Yoshizumi, M.; Chayama, K.; Higashi, Y. The effects of different intensities of acute exercise on systemic hemodynamics and forearm vascular function in humans. Am. J. Hypertens. 2007, 20, 825–830.
- Sessa, W.C.; Pritchard, K.; Seyedi, N.; Wang, J.; Hintze, T.H. Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ. Res. 1994, 74, 349–353.
- Higashi, Y. Assessment of endothelial function: History, methodological aspects, and clinical perspectives. Int. Heart. J. 2015, 56, 125–134.
- Tanaka, A.; Tomiyama, H.; Maruhashi, T.; Matsuzawa, Y.; Miyoshi, T.; Kabutoya, T.; Kario, K.; Sugiyama, S.; Munakata, M.; Uto, H.; et al. Physiological diagnosis criteria for vascular failure. Hypertension 2018, 72, 1060–1071.
- Zhang, F.; Wang, C.; Wang, H.; Lu, M.; Li, Y.; Feng, H.; Lin, J.; Yuan, Z.; Wang, X. Ox-LDL promotes migration and adhesion of bone marrow-derived mesenchymal stem cells via regulation of MCP-1 expression. Mediators. Inflamm. 2013, 2013, 691023.
- Zang, Y.H.; Chen, D.; Zhou, B.; Chen, A.D.; Wang, J.J.; Gao, X.Y.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Zhu, G.Q. FNDC5 inhibits foam cell formation and monocyte adhesion in vascular smooth muscle cells via suppressing NFκB-mediated NLRP3 upregulation. Vascul. Pharmacol. 2019, 121, 106579.
- Chioslm, G.M.; Steinberg, D. The oxidative modification hypothesis of atherogenesis: An overview. Free. Radic. Biol. Med. 2000, 28, 1815–1826.
- Tai, S.C.; Robb, G.B.; Marsden, P.A. Endothelial nitric oxide synthase: A new paradigm for gene regulation in the injured blood vessel. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 405–412.
- Zhang, J.; Ren, S.; Shen, G.X. Glycation amplifies lipoprotein(a)-induced alterations in the generation of fibrinolytic regulators from human vascular endothelial cells. Atherosclerosis 2000, 150, 299–308.
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77.
- Kattoor, A.J.; Goel, A.; Mehta, J.L. LOX-1: Regulation, Signaling and Its Role in Atherosclerosis. Antioxidants 2019, 8, 218.
- Calvier, L.; Herz, J.; Hansmann, G. Interplay of Low-Density Lipoprotein Receptors, LRPs, and Lipoproteins in Pulmonary Hypertension. JACC Basic Transl. Sci. 2022, 7, 164–180.
- Yamashita, N.; Hoshida, S.; Otsu, K.; Asahi, M.; Kuzuya, T.; Hori, M. Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation. J. Exp. Med. 1999, 189, 1699–1706.
- Faraci, F.M.; Didion, S.P. Vascular protection: Superoxide dismutase isoforms in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1367–1373.
- Babior, B.M. NADPH oxidase: An update. Blood 1999, 93, 1464–1476.
- Heyworth, P.G.; Curnutte, J.T.; Nauseef, W.M.; Volpp, B.D.; Pearson, D.W.; Clark, R.A. Neutrofil NADPH oxidase assembly: Translocation of p47phox and p67phox requires the interaction between p47phox and cytochrome b558. J. Clin. Investig. 1991, 87, 352–356.
- Ushio-Fukai, M.; Zafari, A.M.; Fukai, T.; Nishizaka, N.; Griendling, K.K. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J. Biol. Chem. 1996, 271, 23317–23321.
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin II. Circ. Res. 2002, 90, 1205–1213.
- Salazar, G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int. J. Mol. Sci. 2018, 19, 1327.
- Paravicini, T.M.; Touyz, R.M. Redox signaling in hypertension. Cardiovasc. Res. 2006, 71, 247–258.
- Madamanchi, N.R.; Vendrov, A.; Runge, M.S. Oxidative stress and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 29–38.
More
Information
Subjects:
Physics, Applied
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.7K
Revisions:
2 times
(View History)
Update Date:
03 Feb 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No