 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alessandra Galli | -- | 3317 | 2023-02-02 10:05:14 | | | |
| 2 | Rita Xu | -57 word(s) | 3260 | 2023-02-02 10:36:33 | | |
Video Upload Options
Cholesterol, an essential component of the cellular membranes, exhibits multiple structural and functional roles, orchestrating a wide range of signalling pathways and cellular functions. The pancreatic β-cells rely on cholesterol for their survival, proliferation, and functional maturation. Intracellular cholesterol imbalance is a tremendous burden to β-cells as this condition disables the cells from adequately delivering the sterol to its final destinations, resulting in altered plasma membrane organization, impaired mitochondrial activity, reduced insulin granule maturation, and glucose-stimulated insulin secretion. To maintain cholesterol homeostasis, β-cells have evolved a sophisticated machinery that not only controls the lipid biosynthesis or influx/efflux but also its redistribution among the different organelles. The mechanisms by which β-cells sense and shuttle the lipids across different membrane compartments are still largely unexplored.
1. Introduction
2. Insulin Biosynthesis, Sorting and Secretion
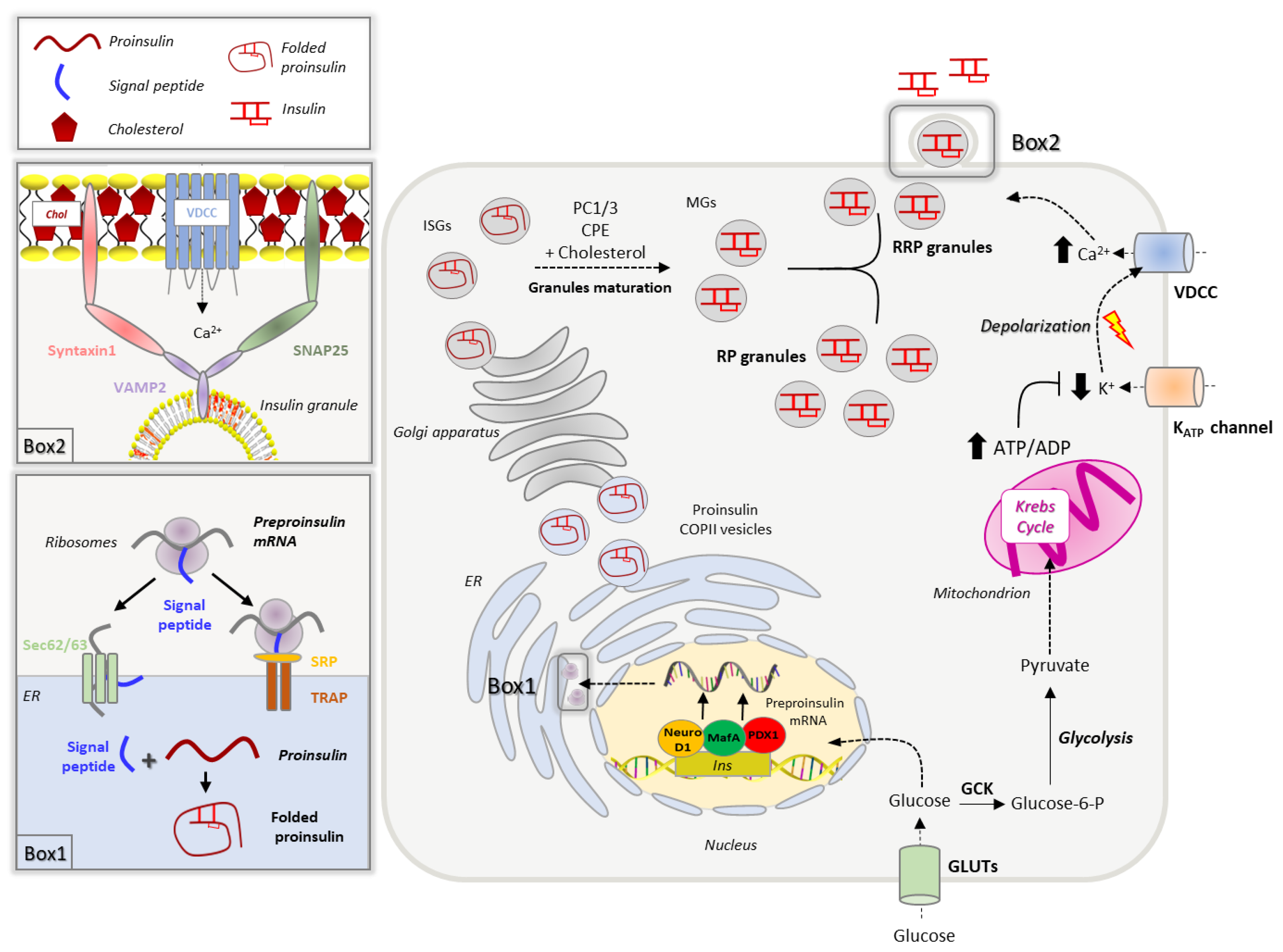
2.1. Insulin Biogenesis, Processing and Storage into Secretory Granules
2.2. Insulin Secretion
3. Cholesterol Biosynthesis, Homeostasis and Turnover
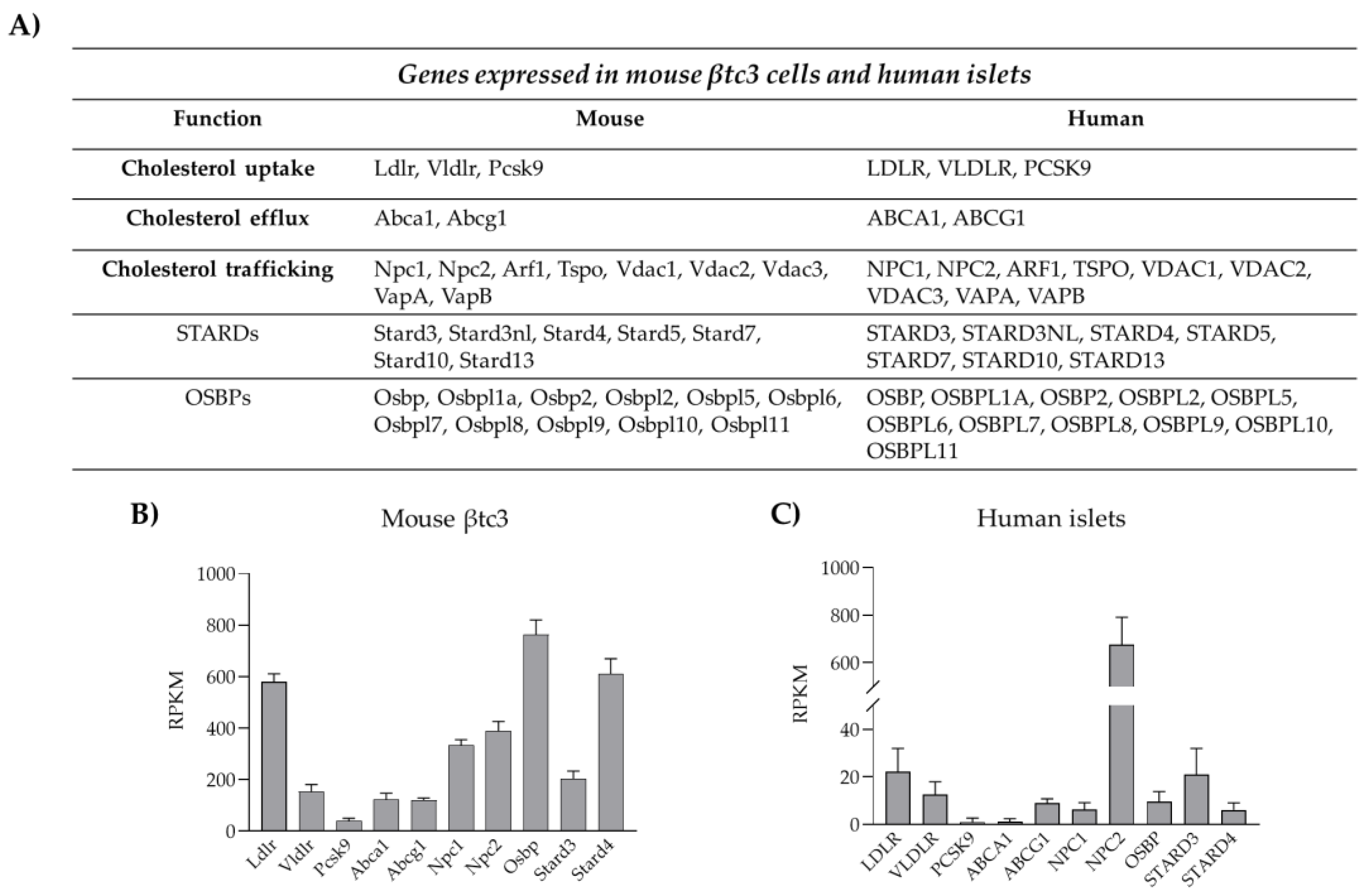
3.1. Cholesterol Biosynthesis
3.2. Cholesterol Uptake
3.3. Cholesterol Esterification and Efflux
References
- Dong, H.; Morral, N.; McEvoy, R.; Meseck, M.; Thung, S.N.; Woo, S.L. Hepatic Insulin Expression Improves Glycemic Control in Type 1 Diabetic Rats. Diabetes Res. Clin. Pract. 2001, 52, 153–163.
- Csajbók, É.A.; Tamás, G. Cerebral Cortex: A Target and Source of Insulin? Diabetologia 2016, 59, 1609–1615.
- Ashcroft, F.M.; Rorsman, P. Diabetes Mellitus and the β Cell: The Last Ten Years. Cell 2012, 148, 1160–1171.
- Rosengren, A.H.; Braun, M.; Mahdi, T.; Andersson, S.A.; Travers, M.E.; Shigeto, M.; Zhang, E.; Almgren, P.; Ladenvall, C.; Axelsson, A.S.; et al. Reduced Insulin Exocytosis in Human Pancreatic β-Cells with Gene Variants Linked to Type 2 Diabetes. Diabetes 2012, 61, 1726–1733.
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global Estimates of Diabetes Prevalence for 2017 and Projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281.
- Taskinen, M.-R.; Borén, J. New Insights into the Pathophysiology of Dyslipidemia in Type 2 Diabetes. Atherosclerosis 2015, 239, 483–495.
- Gregg, E.W.; Gu, Q.; Cheng, Y.J.; Venkat Narayan, K.M.; Cowie, C.C. Mortality Trends in Men and Women with Diabetes, 1971 to 2000. Ann. Intern. Med. 2007, 147, 149.
- Perego, C.; Da Dalt, L.; Pirillo, A.; Galli, A.; Catapano, A.L.; Norata, G.D. Cholesterol Metabolism, Pancreatic β-Cell Function and Diabetes. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2019, 1865, 2149–2156.
- Dirkx, R., Jr.; Solimena, M. Cholesterol-Enriched Membrane Rafts and Insulin Secretion: Membrane Rafts and Insulin Secretion. J. Diabetes Investig. 2012, 3, 339–346.
- Hao, M.; Head, W.S.; Gunawardana, S.C.; Hasty, A.H.; Piston, D.W. Direct Effect of Cholesterol on Insulin Secretion. Diabetes 2007, 56, 2328–2338.
- Bogan, J.S.; Xu, Y.; Hao, M. Cholesterol Accumulation Increases Insulin Granule Size and Impairs Membrane Trafficking: Cholesterol in Insulin Granule Regulation. Traffic 2012, 13, 1466–1480.
- Fryirs, M.; Barter, P.J.; Rye, K.-A. Cholesterol Metabolism and Pancreatic β-Cell Function. Curr. Opin. Lipidol. 2009, 20, 159–164.
- Kruit, J.K.; Wijesekara, N.; Fox, J.E.M.; Dai, X.-Q.; Brunham, L.R.; Searle, G.J.; Morgan, G.P.; Costin, A.J.; Tang, R.; Bhattacharjee, A.; et al. Islet Cholesterol Accumulation Due to Loss of ABCA1 Leads to Impaired Exocytosis of Insulin Granules. Diabetes 2011, 60, 3186–3196.
- Kruit, J.K.; Brunham, L.R.; Verchere, C.B.; Hayden, M.R. HDL and LDL Cholesterol Significantly Influence β-Cell Function in Type 2 Diabetes Mellitus. Curr. Opin. Lipidol. 2010, 21, 178–185.
- Kojima, I.; Medina, J.; Nakagawa, Y. Role of the Glucose-Sensing Receptor in Insulin Secretion. Diabetes Obes. Metab. 2017, 19, 54–62.
- Rorsman, P.; Renström, E. Insulin Granule Dynamics in Pancreatic Beta Cells. Diabetologia 2003, 46, 1029–1045.
- Rutter, G.A. Insulin Secretion: Feed-Forward Control of Insulin Biosynthesis? Curr. Biol. 1999, 9, R443–R445.
- Dumonteil, E.; Philippe, J. Insulin Gene: Organisation, Expression and Regulation. Diabetes Metab. 1996, 22, 164–173.
- Inada, A.; Seino, Y. The regulation of insulin gene transcription by transcription factors. Nihon Rinsho 2002, 60 (Suppl. S7), 159–164.
- Melloul, D.; Cerasi, E. Transcription of the Insulin Gene: Towards Defining the Glucose-Sensitive Cis-Element and Trans-Acting Factors. Diabetologia 1994, 37 (Suppl. S2), S3–S10.
- Someya, Y.; Ishida, H.; Seino, Y. Insulin gene: Gene organization and its regulatory mechanism of transcription. Nihon Rinsho 1997, 55, 73–78.
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes. Curr. Diabetes Rev. 2013, 9, 25–53.
- Kataoka, K.; Han, S.; Shioda, S.; Hirai, M.; Nishizawa, M.; Handa, H. MafA Is a Glucose-Regulated and Pancreatic Beta-Cell-Specific Transcriptional Activator for the Insulin Gene. J. Biol. Chem. 2002, 277, 49903–49910.
- Matsuoka, T.; Zhao, L.; Artner, I.; Jarrett, H.W.; Friedman, D.; Means, A.; Stein, R. Members of the Large Maf Transcription Family Regulate Insulin Gene Transcription in Islet Beta Cells. Mol. Cell Biol. 2003, 23, 6049–6062.
- Raum, J.C.; Gerrish, K.; Artner, I.; Henderson, E.; Guo, M.; Sussel, L.; Schisler, J.C.; Newgard, C.B.; Stein, R. FoxA2, Nkx2.2, and PDX-1 Regulate Islet β-Cell-Specific MafA Expression through Conserved Sequences Located between Base Pairs −8118 and −7750 Upstream from the Transcription Start Site. Mol. Cell Biol. 2006, 26, 5735–5743.
- Han, S.; Aramata, S.; Yasuda, K.; Kataoka, K. MafA Stability in Pancreatic β Cells Is Regulated by Glucose and Is Dependent on Its Constitutive Phosphorylation at Multiple Sites by Glycogen Synthase Kinase 3. Mol. Cell Biol. 2007, 27, 6593–6605.
- Liu, M.; Huang, Y.; Xu, X.; Li, X.; Alam, M.; Arunagiri, A.; Haataja, L.; Ding, L.; Wang, S.; Itkin-Ansari, P.; et al. Normal and Defective Pathways in Biogenesis and Maintenance of the Insulin Storage Pool. J. Clin. Investig. 2021, 131, e142240.
- Wang, S.Y.; Halban, P.A.; Rowe, J.W. Effects of Aging on Insulin Synthesis and Secretion. Differential Effects on Preproinsulin Messenger RNA Levels, Proinsulin Biosynthesis, and Secretion of Newly Made and Preformed Insulin in the Rat. J. Clin. Investig. 1988, 81, 176–184.
- Akopian, D.; Shen, K.; Zhang, X.; Shan, S. Signal Recognition Particle: An Essential Protein-Targeting Machine. Annu. Rev. Biochem. 2013, 82, 693–721.
- Egea, P.F.; Stroud, R.M.; Walter, P. Targeting Proteins to Membranes: Structure of the Signal Recognition Particle. Curr. Opin. Struct. Biol. 2005, 15, 213–220.
- Guo, H.; Sun, J.; Li, X.; Xiong, Y.; Wang, H.; Shu, H.; Zhu, R.; Liu, Q.; Huang, Y.; Madley, R.; et al. Positive Charge in the N-Region of the Signal Peptide Contributes to Efficient Post-Translational Translocation of Small Secretory Preproteins. J. Biol. Chem. 2018, 293, 1899–1907.
- Liu, M.; Weiss, M.A.; Arunagiri, A.; Yong, J.; Rege, N.; Sun, J.; Haataja, L.; Kaufman, R.J.; Arvan, P. Biosynthesis, Structure, and Folding of the Insulin Precursor Protein. Diabetes Obes. Metab. 2018, 20 (Suppl. S2), 28–50.
- Fons, R.D.; Bogert, B.A.; Hegde, R.S. Substrate-Specific Function of the Translocon-Associated Protein Complex during Translocation across the ER Membrane. J. Cell Biol. 2003, 160, 529–539.
- Lakkaraju, A.K.K.; Thankappan, R.; Mary, C.; Garrison, J.L.; Taunton, J.; Strub, K. Efficient Secretion of Small Proteins in Mammalian Cells Relies on Sec62-Dependent Posttranslational Translocation. Mol. Biol. Cell 2012, 23, 2712–2722.
- Bachar-Wikstrom, E.; Wikstrom, J.D.; Ariav, Y.; Tirosh, B.; Kaiser, N.; Cerasi, E.; Leibowitz, G. Stimulation of Autophagy Improves Endoplasmic Reticulum Stress–Induced Diabetes. Diabetes 2013, 62, 1227–1237.
- Cunningham, C.N.; Williams, J.M.; Knupp, J.; Arunagiri, A.; Arvan, P.; Tsai, B. Cells Deploy a Two-Pronged Strategy to Rectify Misfolded Proinsulin Aggregates. Mol. Cell 2019, 75, 442–456.e4.
- Kadowaki, H.; Nagai, A.; Maruyama, T.; Takami, Y.; Satrimafitrah, P.; Kato, H.; Honda, A.; Hatta, T.; Natsume, T.; Sato, T.; et al. Pre-Emptive Quality Control Protects the ER from Protein Overload via the Proximity of ERAD Components and SRP. Cell Rep. 2015, 13, 944–956.
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and Endoplasmic Reticulum Stress in β-Cell Dysfunction in Diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54.
- Mori, K. Tripartite Management of Unfolded Proteins in the Endoplasmic Reticulum. Cell 2000, 101, 451–454.
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes Mellitus and Exocrine Pancreatic Dysfunction in Perk−/− Mice Reveals a Role for Translational Control in Secretory Cell Survival. Mol. Cell 2001, 7, 1153–1163.
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational Control Is Required for the Unfolded Protein Response and In Vivo Glucose Homeostasis. Mol. Cell 2001, 7, 1165–1176.
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The EIF2 Kinase PERK and the Integrated Stress Response Facilitate Activation of ATF6 during Endoplasmic Reticulum Stress. MBoC 2011, 22, 4390–4405.
- Zhang, K.; Kaufman, R.J. The Unfolded Protein Response: A Stress Signaling Pathway Critical for Health and Disease. Neurology 2006, 66, S102–S109.
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799.
- Yang, H.; Niemeijer, M.; van de Water, B.; Beltman, J.B. ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. iScience 2020, 23, 100860.
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 Activated by Proteolysis Binds in the Presence of NF-Y (CBF) Directly to the Cis -Acting Element Responsible for the Mammalian Unfolded Protein Response. Mol. Cell Biol. 2000, 20, 6755–6767.
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The Unfolded Protein Response Signals through High-Order Assembly of Ire1. Nature 2009, 457, 687–693.
- Arunagiri, A.; Haataja, L.; Pottekat, A.; Pamenan, F.; Kim, S.; Zeltser, L.M.; Paton, A.W.; Paton, J.C.; Tsai, B.; Itkin-Ansari, P.; et al. Proinsulin Misfolding Is an Early Event in the Progression to Type 2 Diabetes. eLife 2019, 8, e44532.
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P.; Liu, M. Proinsulin Misfolding and Endoplasmic Reticulum Stress during the Development and Progression of Diabetes. Mol. Asp. Med. 2015, 42, 105–118.
- Miyake, M.; Sobajima, M.; Kurahashi, K.; Shigenaga, A.; Denda, M.; Otaka, A.; Saio, T.; Sakane, N.; Kosako, H.; Oyadomari, S. Identification of an Endoplasmic Reticulum Proteostasis Modulator That Enhances Insulin Production in Pancreatic β Cells. Cell Chem. Biol. 2022, 29, 996–1009.e9.
- Hutton, J.C. Insulin Secretory Granule Biogenesis and the Proinsulin-Processing Endopeptidases. Diabetologia 1994, 37 (Suppl. S2), S48–S56.
- Jo, S.; Lockridge, A.; Alejandro, E.U. EIF4G1 and Carboxypeptidase E Axis Dysregulation in O-GlcNAc Transferase–Deficient Pancreatic β-Cells Contributes to Hyperproinsulinemia in Mice. J. Biol. Chem. 2019, 294, 13040–13050.
- Ramzy, A.; Asadi, A.; Kieffer, T.J. Revisiting Proinsulin Processing: Evidence That Human β-Cells Process Proinsulin With Prohormone Convertase (PC) 1/3 but Not PC2. Diabetes 2020, 69, 1451–1462.
- Arvan, P.; Castle, D. Sorting and Storage during Secretory Granule Biogenesis: Looking Backward and Looking Forward. Biochem. J. 1998, 332 Pt 3, 593–610.
- Traub, L.M.; Kornfeld, S. The Trans-Golgi Network: A Late Secretory Sorting Station. Curr. Opin. Cell Biol. 1997, 9, 527–533.
- Orci, L.; Montesano, R.; Meda, P.; Malaisse-Lagae, F.; Brown, D.; Perrelet, A.; Vassalli, P. Heterogeneous Distribution of Filipin--Cholesterol Complexes across the Cisternae of the Golgi Apparatus. Proc. Natl. Acad. Sci. USA 1981, 78, 293–297.
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane Lipids: Where They Are and How They Behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124.
- Westhead, E.W. Lipid Composition and Orientation in Secretory Vesicles. Ann. N. Y. Acad. Sci. 1987, 493, 92–100.
- Dodson, G.; Steiner, D. The Role of Assembly in Insulin’s Biosynthesis. Curr. Opin. Struct. Biol. 1998, 8, 189–194.
- Olofsson, C.S.; Göpel, S.O.; Barg, S.; Galvanovskis, J.; Ma, X.; Salehi, A.; Rorsman, P.; Eliasson, L. Fast Insulin Secretion Reflects Exocytosis of Docked Granules in Mouse Pancreatic B-Cells. Pflug. Arch. 2002, 444, 43–51.
- Henquin, J.C. Triggering and Amplifying Pathways of Regulation of Insulin Secretion by Glucose. Diabetes 2000, 49, 1751–1760.
- Henquin, J.-C.; Nenquin, M.; Stiernet, P.; Ahren, B. In Vivo and in Vitro Glucose-Induced Biphasic Insulin Secretion in the Mouse: Pattern and Role of Cytoplasmic Ca2+ and Amplification Signals in Beta-Cells. Diabetes 2006, 55, 441–451.
- De Vos, A.; Heimberg, H.; Quartier, E.; Huypens, P.; Bouwens, L.; Pipeleers, D.; Schuit, F. Human and Rat Beta Cells Differ in Glucose Transporter but Not in Glucokinase Gene Expression. J. Clin. Investig. 1995, 96, 2489–2495.
- McCulloch, L.J.; van de Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) Is Not the Principal Glucose Transporter in Human Pancreatic Beta Cells: Implications for Understanding Genetic Association Signals at This Locus. Mol. Genet. Metab. 2011, 104, 648–653.
- Seino, S.; Sugawara, K.; Yokoi, N.; Takahashi, H. β-Cell Signalling and Insulin Secretagogues: A Path for Improved Diabetes Therapy. Diabetes Obes. Metab. 2017, 19, 22–29.
- Ammälä, C.; Eliasson, L.; Bokvist, K.; Larsson, O.; Ashcroft, F.M.; Rorsman, P. Exocytosis Elicited by Action Potentials and Voltage-Clamp Calcium Currents in Individual Mouse Pancreatic B-Cells. J. Physiol. 1993, 472, 665–688.
- Wollheim, C.B.; Sharp, G.W. Regulation of Insulin Release by Calcium. Physiol. Rev. 1981, 61, 914–973.
- Bruns, D.; Jahn, R. Molecular Determinants of Exocytosis. Pflug. Arch. 2002, 443, 333–338.
- Xia, F.; Gao, X.; Kwan, E.; Lam, P.P.L.; Chan, L.; Sy, K.; Sheu, L.; Wheeler, M.B.; Gaisano, H.Y.; Tsushima, R.G. Disruption of Pancreatic β-Cell Lipid Rafts Modifies Kv2.1 Channel Gating and Insulin Exocytosis. J. Biol. Chem. 2004, 279, 24685–24691.
- Dolai, S.; Xie, L.; Zhu, D.; Liang, T.; Qin, T.; Xie, H.; Kang, Y.; Chapman, E.R.; Gaisano, H.Y. Synaptotagmin-7 Functions to Replenish Insulin Granules for Exocytosis in Human Islet β-Cells. Diabetes 2016, 65, 1962–1976.
- Huang, C.; Walker, E.M.; Dadi, P.K.; Hu, R.; Xu, Y.; Zhang, W.; Sanavia, T.; Mun, J.; Liu, J.; Nair, G.G.; et al. Synaptotagmin 4 Regulates Pancreatic β Cell Maturation by Modulating the Ca2+ Sensitivity of Insulin Secretion Vesicles. Dev. Cell 2018, 45, 347–361.e5.
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.R.M. Metabolic Signaling in Fuel-Induced Insulin Secretion. Cell Metab. 2013, 18, 162–185.
- Di Cairano, E.S.; Moretti, S.; Marciani, P.; Sacchi, V.F.; Castagna, M.; Davalli, A.; Folli, F.; Perego, C. Neurotransmitters and Neuropeptides: New Players in the Control of Islet of Langerhans’ Cell Mass and Function. J. Cell Physiol. 2016, 231, 756–767.
- Churchward, M.A.; Coorssen, J.R. Cholesterol, Regulated Exocytosis and the Physiological Fusion Machine. Biochem. J. 2009, 423, 1–14.
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.-M.; Andréasson, A.-C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607.
- Shao, W.; Espenshade, P.J. Lipids. Cholesterol Synthesis and Regulation. In Encyclopedia of Biological Chemistry III; Elsevier: Amsterdam, The Netherlands, 2021; pp. 732–738. ISBN 978-0-12-822040-5.
- Ishikawa, M.; Iwasaki, Y.; Yatoh, S.; Kato, T.; Kumadaki, S.; Inoue, N.; Yamamoto, T.; Matsuzaka, T.; Nakagawa, Y.; Yahagi, N.; et al. Cholesterol Accumulation and Diabetes in Pancreatic β-Cell-Specific SREBP-2 Transgenic Mice: A New Model for Lipotoxicity. J. Lipid Res. 2008, 49, 2524–2534.
- Grupping, A.Y.; Cnop, M.; Van Schravendijk, C.F.H.; Hannaert, J.C.; Van Berkel, T.J.C.; Pipeleers, D.G. Low Density Lipoprotein Binding and Uptake by Human and Rat Islet β Cells. Endocrinology 1997, 138, 4064–4068.
- Fujino, T.; Asaba, H.; Kang, M.-J.; Ikeda, Y.; Sone, H.; Takada, S.; Kim, D.-H.; Ioka, R.X.; Ono, M.; Tomoyori, H.; et al. Low-Density Lipoprotein Receptor-Related Protein 5 (LRP5) Is Essential for Normal Cholesterol Metabolism and Glucose-Induced Insulin Secretion. Proc. Natl. Acad. Sci. USA 2003, 100, 229–234.
- d Oliveira, R.B.; Carvalho, C.P.d.F.; Polo, C.C.; Dorighello, G.d.G.; Boschero, A.C.; d Oliveira, H.C.F.; Collares-Buzato, C.B. Impaired Compensatory Beta-Cell Function and Growth in Response to High-Fat Diet in LDL Receptor Knockout Mice. Int. J. Exp. Pathol. 2014, 95, 296–308.
- Ye, R.; Gordillo, R.; Shao, M.; Onodera, T.; Chen, Z.; Chen, S.; Lin, X.; SoRelle, J.A.; Li, X.; Tang, M.; et al. Intracellular Lipid Metabolism Impairs β Cell Compensation during Diet-Induced Obesity. J. Clin. Investig. 2018, 128, 1178–1189.
- Norata, G.D.; Tibolla, G.; Catapano, A.L. Targeting PCSK9 for Hypercholesterolemia. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 273–293.
- Seidah, N.; Mayer, G.; Zaid, A.; Rousselet, E.; Nassoury, N.; Poirier, S.; Essalmani, R.; Prat, A. The Activation and Physiological Functions of the Proprotein Convertases. Int. J. Biochem. Cell Biol. 2008, 40, 1111–1125.
- Cariou, B.; Si-Tayeb, K.; Le May, C. Role of PCSK9 beyond Liver Involvement. Curr. Opin. Lipidol. 2015, 26, 155–161.
- Da Dalt, L.; Ruscica, M.; Bonacina, F.; Balzarotti, G.; Dhyani, A.; Di Cairano, E.; Baragetti, A.; Arnaboldi, L.; De Metrio, S.; Pellegatta, F.; et al. PCSK9 Deficiency Reduces Insulin Secretion and Promotes Glucose Intolerance: The Role of the Low-Density Lipoprotein Receptor. Eur. Heart J. 2019, 40, 357–368.
- Marku, A.; Da Dalt, L.; Galli, A.; Dule, N.; Corsetto, P.; Rizzo, A.M.; Moregola, A.; Uboldi, P.; Bonacina, F.; Marciani, P.; et al. Pancreatic PCSK9 Controls the Organization of the β-Cell Secretory Pathway via LDLR-Cholesterol Axis. Metabolism 2022, 136, 155291.
- Mbikay, M.; Sirois, F.; Mayne, J.; Wang, G.-S.; Chen, A.; Dewpura, T.; Prat, A.; Seidah, N.G.; Chretien, M.; Scott, F.W. PCSK9-Deficient Mice Exhibit Impaired Glucose Tolerance and Pancreatic Islet Abnormalities. FEBS Lett. 2010, 584, 701–706.
- Ramin-Mangata, S.; Thedrez, A.; Nativel, B.; Diotel, N.; Blanchard, V.; Wargny, M.; Aguesse, A.; Billon-Crossouard, S.; Vindis, C.; Le May, C.; et al. Effects of Proprotein Convertase Subtilisin Kexin Type 9 Modulation in Human Pancreatic Beta Cells Function. Atherosclerosis 2021, 326, 47–55.
- Tong, X.; Liu, S.; Stein, R.; Imai, Y. Lipid Droplets’ Role in the Regulation of β-Cell Function and β-Cell Demise in Type 2 Diabetes. Endocrinology 2022, 163, bqac007.
- Neumann, J.; Rose-Sperling, D.; Hellmich, U.A. Diverse Relations between ABC Transporters and Lipids: An Overview. Biochim. Et Biophys. Acta (BBA)—Biomembr. 2017, 1859, 605–618.
- Oram, J. Tangier Disease and ABCA1. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2000, 1529, 321–330.
- Singaraja, R.R.; Brunham, L.R.; Visscher, H.; Kastelein, J.J.P.; Hayden, M.R. Efflux and Atherosclerosis: The Clinical and Biochemical Impact of Variations in the ABCA1 Gene. ATVB 2003, 23, 1322–1332.
- Sturek, J.M.; Castle, J.D.; Trace, A.P.; Page, L.C.; Castle, A.M.; Evans-Molina, C.; Parks, J.S.; Mirmira, R.G.; Hedrick, C.C. An Intracellular Role for ABCG1-Mediated Cholesterol Transport in the Regulated Secretory Pathway of Mouse Pancreatic β Cells. J. Clin. Investig. 2010, 120, 2575–2589.
- Brunham, L.R.; Kruit, J.K.; Pape, T.D.; Timmins, J.M.; Reuwer, A.Q.; Vasanji, Z.; Marsh, B.J.; Rodrigues, B.; Johnson, J.D.; Parks, J.S.; et al. β-Cell ABCA1 Influences Insulin Secretion, Glucose Homeostasis and Response to Thiazolidinedione Treatment. Nat. Med. 2007, 13, 340–347.
- Tarr, P.T.; Tarling, E.J.; Bojanic, D.D.; Edwards, P.A.; Baldán, Á. Emerging New Paradigms for ABCG Transporters. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2009, 1791, 584–593.
- Gerin, I.; Dolinsky, V.W.; Shackman, J.G.; Kennedy, R.T.; Chiang, S.-H.; Burant, C.F.; Steffensen, K.R.; Gustafsson, J.-Å.; MacDougald, O.A. LXRβ Is Required for Adipocyte Growth, Glucose Homeostasis, and β Cell Function. J. Biol. Chem. 2005, 280, 23024–23031.

