Pancreatic β-cells are responsible for maintaining the blood glucose levels within a very narrow range by synthesizing and secreting the major hypoglycemic hormone, insulin. Despite reports indicating insulin expression in tissues, such as the brain or liver [
1,
2], the pancreatic β-cells remain the major source of insulin biosynthesis and secretion; therefore, their dysfunction and/or death result in decompensated glucose homeostasis and diabetes development [
3,
4]. Diabetes indicates a group of heterogeneous metabolic disorders characterized by fasting hyperglycemia; among them, type 2 diabetes (T2D) deserves particular attention given its exponential growth worldwide; it is expected to reach almost 700 million cases in 2045 [
5]. T2D is often associated with a cluster of lipid abnormalities—which include low HDL (high-density lipoproteins) and increased levels of triglycerides and remnant lipoproteins (LDL, low-density lipoproteins, and VLDL, very low-density lipoproteins)—involved in cholesterol transport [
6]. Diabetic dyslipidemia not only increases the risk of cardiovascular events but also contributes to the β-cells dysfunction [
7,
8]. Indeed, cholesterol can be either “friend or foe” of pancreatic β-cells as appropriate cholesterol levels are necessary for β-cell functions, insulin biogenesis and secretion; however, inadequate cholesterol levels strongly affect the ability of β-cells to synthetize and secrete the proper amount of insulin, while an excess can lead to β-cell demise [
9,
10,
11,
12,
13,
14].
2. Insulin Biosynthesis, Sorting and Secretion
The insulin gene, located on the chromosome 11, codes for a 110-amino acid precursor, called pre-proinsulin, which undergoes multiple enzymatic reactions to yield insulin. Insulin biogenesis and secretion are multi-step processes that are tightly controlled by nutrient availability. Although several nutrients may control insulin secretion, glucose is the well-characterized one [
15,
16]. To sustain β-cell insulin secretion, glucose not only promotes hormone release but also increases the hormone transcription and translation. Interestingly, insulin release begins in seconds after the increase of glucose levels, while mRNA translation and transcription are activated from minutes to hours [
17]. The main steps of insulin production and secretion are summarized in
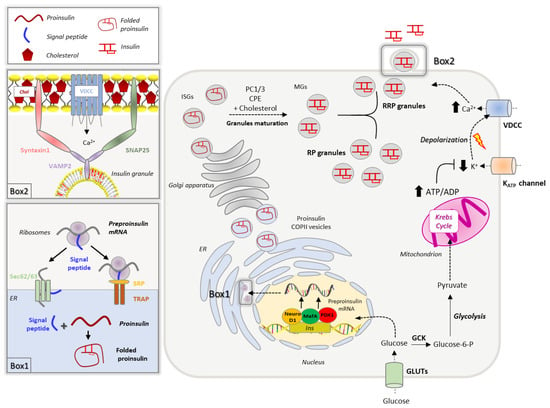
Figure 1.
Figure 1. Insulin biosynthesis and secretion. Extracellular glucose is transported in the β-cells through GLUT transporters (GLUT1 in humans, GLUT2 in rodents). Glucose increase promotes the insulin gene transcription, which is controlled by PDX1, MafA and NeuroD1 transcription factors. The insulin transcript crosses the ER membrane via co-translational (SRP and TRAP-dependent) and post-translational (Sec62/63-dependent) translocation processes. Once in the ER lumen, the pre-proinsulin signal peptide is rapidly cleaved to yield proinsulin that undergoes oxidative folding (Box 1). Newly synthesized proinsulin, loaded into COPII vesicles, is then delivered to the Golgi apparatus, where it is sorted to immature secretory granules (ISGs). Granules maturation is associated with increased cholesterol accumulation in the membrane and proinsulin-to-insulin conversion mediated by prohormone convertases 1/3 (PC1/3) and carboxypeptidase E (CPE). Mature granules (MGs) are sorted into two different pools: the readily releasable pool (RRP) docked to the plasma membrane and the reserve pool (RP). Insulin secretion is promoted by the GCK-mediated phosphorylation of glucose, which is then metabolized via glycolysis and Krebs cycle, leading to an increase of ATP production. The elevation of ATP/ADP ratio promotes the closure of K+ATP channels, membrane depolarization and activation of voltage-gated Ca2+ channels (VDCC). The increase of intracellular Ca2+ triggers the insulin granule fusion with the plasma membrane, a process regulated by the SNARE complex, composed by syntaxin1 and SNAP25 on the plasma membrane and the vesicular protein VAMP2; these multiprotein complexes occur in specific plasma membrane domains enriched in cholesterol (Chol), called lipid rafts (Box 2).
2.1. Insulin Biogenesis, Processing and Storage into Secretory Granules
A network of transcription factors, including PDX-1 (pancreatic and duodenal homeobox), BETA2/NeuroD1 (BETA2/Neurogenic differentiation 1) and MafA (v-maf musculoaponeurotic fibrosarcoma oncogene homologue), promotes insulin gene transcription in response to increased blood glucose by binding to conserved cis-acting regulatory elements (A3, C1, E1 elements) present in the insulin promoter region [
18,
19,
20,
21]. Although MafA can activate the insulin promoter alone by binding to the C element, the tethering of the BETA2/NeuroD1 and PDX1 to the elements E and A [
22] synergically increases the insulin transcription rate [
23,
24,
25]. The stability of the MafA transcription factor and its binding to the regulatory elements of the
Ins promoter is modulated by phosphorylation, in a glucose-dependent manner [
26].
Once synthesized, insulin mRNA binds to specific regulatory elements that escort it to the ribosomes to initiate translation [
27,
28]. Newly synthesized pre-proinsulin (110 amino acids) contains a hydrophobic N-terminal sequence that recognizes cytosolic ribonucleoprotein signal recognition particles (SRPs) facilitating its translocation across the endoplasmic reticulum (ER) membrane [
29,
30]. This process, in which translation is coupled to translocation, was long considered to be the only mechanism responsible for the protein accumulation into the ER; about 5–15% of synthesized pre-proinsulin molecules are not directly translocated into the ER [
31,
32]. These molecules are released in the cytoplasm and rescued through the post-translation translocation route, which is SRP-independent and relies on the specific component of the translocation machinery, as Sec62/63 and the signal sequence receptor TRAP [
32,
33,
34].
Upon delivery to the ER lumen, the signal peptide is rapidly cleaved to yield proinsulin, which undergoes chaperone-mediated folding forming three disulphide bonds. The ER folding machinery meticulously controls proinsulin in order to recognize misfolded or unfolded molecules that are eliminated through ERAD (ER-associated protein degradation)-dependent ubiquitination or ER-macroautophagy [
31,
35,
36,
37]. An accumulation of misfolded proinsulin activates the unfolded protein response (UPR) in order to alleviate the ER load and restore the folding capability [
38]. UPR activation is primarily mediated by the heat shock protein/chaperone BIP (GRP78), which dissociates from and activates PERK (protein kinase R-like ER kinase), IRE1 (interferon response element 1) and ATF6 (activating transcription factor 6), the three major ER stress sensors [
22,
38,
39]. Through the phosphorylation of eIF2α, PERK induces the transcription of CHOP (C/EBP homologous protein) and ATF4, which alleviates the ER load by reducing the pre-proinsulin translation; PERK can also support the expression of ATF6, another key component of the UPR machinery [
40,
41,
42,
43]. ATF6 translocates from the ER to Golgi, where it is cleaved, and migrates to the nucleus where it induces the transcription of CHOP, XBP1 and other chaperones involved in protein folding and degradation [
44,
45,
46]. Protein folding is also supported by the activation of IRE1, which promotes the transcription of genes encoding for folding enzymes [
43,
47]. In healthy β-cells, approximately 20% of proinsulin misfold, resulting in a basal activation of the UPR machinery that continuously restores ER homeostasis. On the other hand, stressed β-cells (as seen in T2D) could exhibit severe ER homeostasis defects that may result in proinsulin misfolding [
48]. When the ER stress levels increase beyond a threshold, either because of excessive protein synthesis demand (and overworked β-cells) or due to over accumulation of misfolded proteins in the ER lumen, the UPR sensors trigger signaling pathways and transcriptional events that induce β-cell dysfunction and death [
38,
49]. A very recent work has revealed that a pharmacological reversal of UPR or protein aggregation in β-cells improved cellular insulin content and release [
50], reinforcing the ER homeostasis-to-insulin biosynthesis link in the pancreatic β-cells.
Folded proinsulin is delivered through COPII vesicles to the Golgi, where a complex set of events coordinates insulin granule formation and maturation [
27]. Upon transport through the Golgi complex, proinsulin, together with its processive enzymes, is loaded into immature secretory granules, characterized by high Ca
2+ levels and low pH, the optimal environment for the activation of endopeptidases. The proteolytical cleavage of proinsulin by prohormone convertase 1/3 (PC1/3) and the removal of the C-terminal domain by carboxypeptidase E (CPE) result in the formation of mature insulin, consisting of A and B chains held by two disulphide bonds [
51,
52,
53]. The cleaved C-terminus, known as C-peptide, remains within the granules and is co-secreted with insulin. Along with the proinsulin-to-insulin conversion, the maturation of insulin granules requires a shift in proteins and lipid composition, which leads to the acquisition of competence for stimulus-dependent secretion [
27,
54]. During transport in the trans-Golgi network (TGN), granules expose their membrane adaptor proteins, a process that facilitates the assembly of the clathrin coat and its targeting to the plasma membrane [
54,
55]. Changes in lipid composition occur as well; indeed, membrane cholesterol progressively increases reaching 35 mol% in the TGN, a concentration similar to that found in the plasma membrane [
56,
57,
58]. This cholesterol enrichment is required for the sorting of endopeptidases and adaptor proteins within the forming granules [
9] and to confer the membrane curvature necessary for the budding of granules from the TGN [
11]. Then granules progressively mature and acquire a dense core formed by Zn
2+–insulin crystals, while unwanted cargoes and membrane proteins are retrieved back to the TGN by retrograde transport [
59].
Once mature, insulin granules are distributed into two different pools, a small fraction (1–5%), known as the ready releasable pool (RRP), is docked to the plasma membrane and is released on demand without any further modification, while the remaining granules (95–99%) belong to the reserve pool (RP), which must undergo a series of preparatory reactions before acquiring release competence [
16,
60].
2.2. Insulin Secretion
Insulin secretion shows a canonical biphasic pattern consisting of a rapid and transient first phase due to the release of RRP granules, followed by a prolonged second phase [
60,
61,
62]. The first phase begins when blood glucose levels exceed 100 mg/dL, which leads to its internalization through GLUT1 transporters (GLUT2 in rodents) into the β-cells [
63,
64]. Once in the cytoplasm, glucose is phosphorylated by the glucokinase (GCK), enzyme that functions as a glucose sensor [
8,
65]. At low intracellular glucose levels, GCK is inactive and associated with insulin granules through the interaction with nNOS (dimeric neuronal nitric oxide synthase). The increase of intracellular glucose promotes the dissociation of GCK from nNOS and its translocation into the cytoplasm, where GCK phosphorylates glucose to generate glucose-6-phosphate that enters into the glycolytic pathway [
16]. Pyruvate, the final product of glycolysis, is then oxidized via the Krebs cycle into the mitochondria to produce ATP. The increased ATP/ADP ratio induces the closure of K
+ATP channels leading to cellular depolarization, which in turn, triggers the opening of L-type voltage gated Ca
2+ channels (VDCC) [
66,
67]. The rapid calcium influx promotes the fusion of insulin granules with the plasma membrane, a process that involves the spatial reorganization of multiprotein machinery known as the SNARE (soluble-N-ethyl-maleimide sensitive factor-attachment protein receptor) complex. In mature pancreatic β-cells, the main components of SNAREs are syntaxin 1 and SNAP-25 (soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein receptor) on the plasma membrane and the vesicular protein VAMP2 (vesicle-associated membrane protein 2), also known as synaptobrevin [
68]. The SNARE complex acts as a zipper, tethering the granules to the plasma membrane, and ensuring Ca
2+ entry in the proximity of docked granules. Indeed, syntaxin1 and SNAP-25 bind the second and third intracellular loops of the L-type calcium channels, keeping them in close contact with the secretory granules [
16,
61,
68]. The spatial organization of the SNARE complex is guaranteed by their localization in specific domains within the plasma membrane, known as lipid rafts, where L-type Ca
2+ and K
+ATP channels are also enriched [
69]. Alongside SNAREs, Synaptotagmin (Syt) family proteins play a versatile role in insulin exocytosis. While Syt-7 has been recognized as aiding granule recruitment to the plasma membrane upon glucose stimulus [
70], Syt-4 has been indicated to regulate the RRP insulin granule quantities as well as the localization of the granules to the plasma membrane [
71]. The second phase of insulin secretion relies on the mobilization of granules belonging to the reserve pool (RP) and can be maintained for several hours until glucose levels remain elevated [
62]. Unlike the first phase, prolonged insulin release is mainly stimulated by metabolic intermediates, as acetyl-CoA, ATP, NADPH, and α-ketoglutarate/glutamate, that amplify the glucose-induced insulin secretion [
72]. Besides glucose, other substrates including amino acids, lipids and paracrine signals can control insulin secretion using parallel mechanisms [
73].
3. Cholesterol Biosynthesis, Homeostasis and Turnover
Insulin biosynthesis, sorting and secretion are affected by cholesterol levels as the lipid not only represents a fuel for the β-cells but also a key constituent of their membrane domains. Thanks to its rigid and planar structure, cholesterol contributes to the physical properties and organization of cellular membranes affecting cellular permeability, signaling activity, organelles architecture and vesicular trafficking [
12,
74]. Given the key structural and functional roles of cholesterol, pancreatic β-cells are equipped with sophisticated machinery able to control cholesterol synthesis, uptake and efflux, as well as its intracellular distribution [
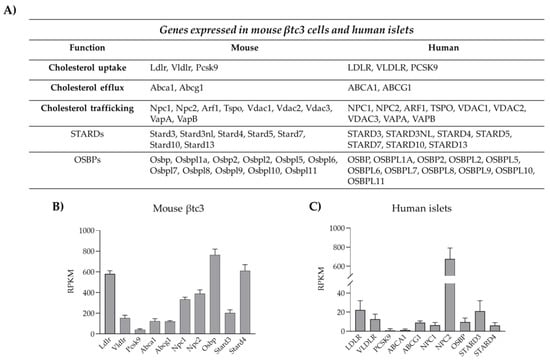
8] (
Figure 2).
Figure 2. Machinery involved in cholesterol homeostasis and trafficking in pancreatic β-cells. (
A) Genes involved in cholesterol uptake/efflux and lipid trafficking expressed in mouse βtc3 cell line and human isolated islets of Langerhans. (
B) RNA-seq data of genes involved in cholesterol homeostasis and expressed in mouse βtc3 cells (unpublished data). Data are expressed as reads per kilobase of transcript per million (RPKM) and are mean ± SD (n = 5 samples). (
C) RNA-seq of genes involved in cholesterol homeostasis and expressed in human islets of Langerhans. Data are expressed as reads per kilobase of transcript per million (RPKM) and are mean ± SD (n = 6 healthy donors of varying BMI and age). The analysis on human islets was performed using the dataset published by Segerstolpe et al., 2016 [
75].
3.1. Cholesterol Biosynthesis
Pancreatic β-cells synthetize cholesterol in the ER through the mevalonate pathway, a complex cascade of enzymatic reactions starting with the condensation of acetyl-coenzyme A (CoA) and acetoacetyl-CoA to form 3-hydroxy-3-methylglutaryl-CoA (HMGCoA). This reaction is catalyzed by hydroxymethylglutaryl-CoA reductase (HMGCoAR), which represents the limiting enzyme for cholesterol synthesis [
76]. The transcription of HMGCoAR is controlled by the transcription factor SREBP2 (sterol-responsive element binding protein 2), which is active only when cholesterol levels are low. SREBP2 is present in the inactive form, bound to the SREBP cleavage-activating protein (SCAP), which is the real sensor of sterols in the cell. When cholesterol concentration decreases, the SREBP2 detaches from SCAP and translocates into the nucleus where it binds to sterol regulatory sequences (SREs) present in the HMGCoAR promoter region, favoring its transcription [
8,
77]. The importance of SREBP2-HMAGCoAR axis in β-cell homeostasis was demonstrated by studies in transgenic mice overexpressing SREBP2. Cholesterol accumulation in the transgenic model of in vivo activation of SREBP2 resulted in reduced β-cell mass and insulin-deficient diabetes [
77].
3.2. Cholesterol Uptake
Cholesterol uptake from plasma lipoproteins represents another mechanism for cholesterol accumulation into β-cells. Lipoprotein transport is mediated by receptor systems that selectively recognize and bind LDL (low density lipoprotein), VLDL (very low density lipoprotein) and HDL (high density lipoprotein). Pancreatic β-cells are equipped with different lipoprotein receptors, including the LDL receptor (LDLR), the VLDL receptor (VLDLR), the receptor for oxidized LDL (CD36) and the scavenger B1 receptor (SR-B1), which is selective for HDL [
12,
78]. Unlike the others, LDLR requires co-receptors for its function, as LPR5; the importance of LPR5 for LDLR activity has been demonstrated in LPR5 knock-out mice, which showed reduced hepatic chylomicron clearance, insulin secretion and glucose tolerance [
79]. Moreover, global LDLR-KO mice displayed increased blood cholesterol levels and were susceptible to high-fat diet with purified islets from those showing poor glucose-stimulated insulin secretion [
80]. Likewise, a study using β-cell-specific knockout of LDL receptor-related protein 1 (LRP1) explored the relationships between the type of diet, the LDL receptor function and lipid metabolism [
81]. The majority of studies regarding the impact of dysregulated cholesterol uptake on insulin secretion have been focused on LDLR as it is highly expressed in pancreatic β-cells, as shown in
Figure 2.
Cholesterol uptake is directly regulated by cholesterol availability via modulation of LDLR transcription, a process mediated by SREBP2, which therefore represents the master regulator of cholesterol levels in β-cells [
77]. An alternative mechanism is represented by post-translational regulations of LDLR expression. In the liver, LDLR abundance on the plasma membrane is controlled by the proprotein convertase subtilisin/kexin type 9 (PCSK9), an enzyme that routes LDLR to the lysosomes for degradation, thus preventing its recycling to the plasma membrane [
82,
83]. Very recently, we and others have shown that LDLR expression, and thus cholesterol uptake, are modulated by PCSK9 also in pancreatic β-cells [
84,
85,
86,
87]. PCSK9 full knock-out mice exhibit reduced plasma cholesterol levels, but increased cholesterol content in the islets of Langerhans, as a consequence of increased LDLR expression in β-cells. Mice developed glucose-intolerance that appears to be the result of insulin insufficiency rather than insulin resistance. In accordance, PCSK9 KO mice presented altered islets morphology, along with dysregulated insulin release [
85,
87]. In contrast, mice in which PCSK9 is switched off selectively in the liver exhibited intact glucose metabolism, revealing that pancreatic PCSK9, rather than the hepatic-circulating protein, is involved in the control of LDLR expression in pancreatic islets [
8,
85]. Indeed, PCSK9 can be synthesized and secreted by pancreatic δ- and β-cells and might act locally in paracrine/autocrine manners. We have recently confirmed this hypothesis in a novel animal model where PCSK9 expression was deleted only in PDX1 expressing cells, namely β- and δ-cells; transgenic mice exhibited cholesterol accumulation in the β-cells and defective insulin secretion, confirming that the pancreatic PCSK9-LDLR-cholesterol axis plays a prominent role in the regulation of cholesterol homeostasis in pancreatic islets [
86]. In contrast to these findings, a separate study involving gene silencing or pharmacological inhibition of PCSK9 in human EndoC-βH1 cells suggested that while PCSK9 knockdown or functional inhibition resulted in increased expression of cell surface LDLR, glucose stimulated insulin secretion decrement was not observed, however observing ~50% loss in GSIS when cells were directly exposed to LDL [
88]. Species difference (human vs. mouse) or the stage of β-cell differentiation (embryonic vs. adult) could be likely explanations for the observed phenotype differences. In accordance, expression analysis of cholesterol handling proteins highlighted a great heterogeneity among β-cell lines; thus, it is also possible that distinct compensatory mechanisms in β-cells might set in upon the severe loss of PCSK9. Further investigations are necessary to clarify this point.
3.3. Cholesterol Esterification and Efflux
As excess free cholesterol can be toxic, cells normally convert sterols in cholesteryl esters by the enzyme acyl-coenzyme A:cholesterol acyltransferase (ACAT) 1 and store them in lipid droplets. Little is known about ACAT1 in β-cells, but lipid droplets have been observed in human islets of Langerhans and found to be enriched in aged and T2D islets [
89]. An additional mechanism to remove excess cholesterol is the efflux via the SR-B1 receptor (which mediates both cholesterol uptake and efflux) and ATP-binding cassette (ABC) transporters, which transport cholesterol against gradient through ATP hydrolysis [
12]. ABC transporters belong to five different subfamilies (A–G), which have different cellular distribution and substrate specificity [
90]. Among them, ABCA1 and ABCG1 are the main players in mediating cholesterol efflux, as suggested by the fact that genetic mutations of ABCA1 lead to the Tangier disease, a genetic disorder characterized by cholesterol accumulation in various organs, atherosclerosis and premature coronaropathy [
91,
92]. ABCA1 transports cholesterol specifically to HDL lipoproteins, while ABCG1 mediates cholesterol efflux to non-specific acceptors as HDL, LDL and cyclodextrin [
90]. As in other tissues, ABCA1 and ABCG1 are the major contributors of cholesterol efflux also in pancreatic β-cells as revealed by the increased cholesterol accumulation in pancreatic islets of ABCA1 and ABCG1 knock-out mice [
93,
94]. The expression of ABCA1 and ABCG1 is strictly regulated by the intracellular content of cholesterol and/or oxysterols (oxygenated derivatives of cholesterol). Indeed, these sterols promote the transcription of ABCA1 and ABCG1 functioning as ligand-activators of the LXR-RXR pathway (liver-X-receptor/retinoic acid receptor) [
95]. Accordingly, disruption of the LXR-RXR axis, due to genetic ablation of LXR, causes increased cholesterol accumulation and β-cell dysfunction [
96].