 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Donald C. Vinh | -- | 2801 | 2023-02-01 20:20:14 | | | |
| 2 | Catherine Yang | -4 word(s) | 2797 | 2023-02-02 04:23:25 | | |
Video Upload Options
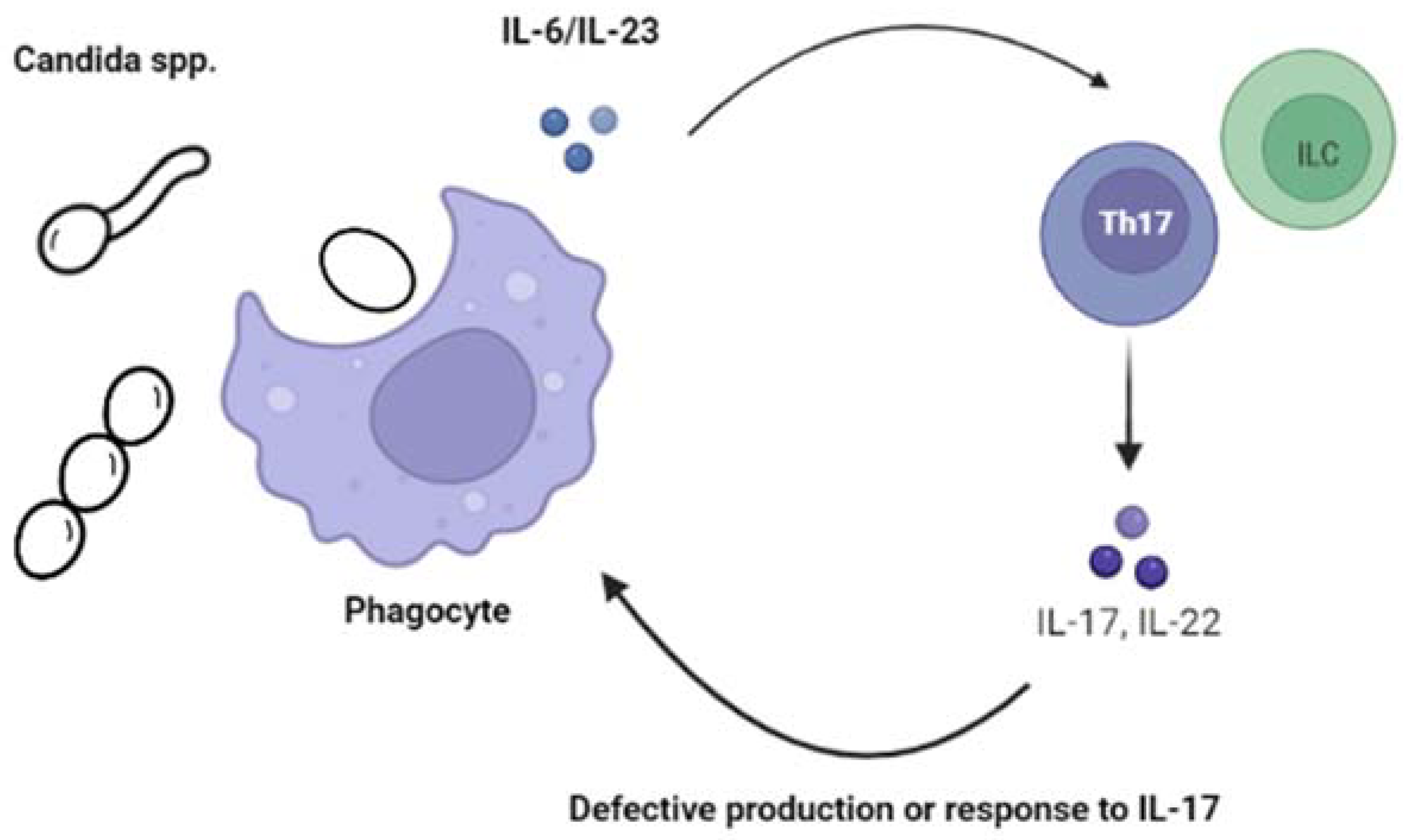
Candida spp. can be found colonizing the skin, oral mucosa, and/or the gastrointestinal and genitourinary tracts in healthy individuals. Most cases of subsequent disease usually emerge from this endogenous microbiota. Although there are now more than 200 species of Candida identified, only a relatively small and yet not clearly defined number (~10–20) have been reported in the context of disease in children and adults. Historically, C. albicans has been, by far, the most commonly isolated species, although the rates of non-albicans species have variably increased globally, probably due to modifications in prophylactic approaches as well as changes in the characteristics of the most vulnerable patient populations, such as preterm neonates and immunosuppressed children. Overall, Candida infections in children can be divided into two main disease presentations: chronic mucocutaneous candidiasis, which can manifest as oropharyngeal candidiasis (OPC, also known as “thrush”), esophagitis, diaper dermatitis, onychomycosis, and/or vulvovaginitis, and invasive candidiasis (IC).
1. Chronic Mucocutaneous Candidiasis (CMC)
2. Invasive Candidiasis (IC)
References
- Smith, B.P.; Steinbach, W.J. Candida Species. In Principles and Practice of Pediatric Infectious Diseases, 6th ed.; Long, S.S., Prober, C.G., Fischer, M., Kimberlin, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Chapter 243.
- Kirkpatrick, C.H. Chronic mucocutaneous candidiasis. Pediatr. Infect. Dis. J. 2001, 20, 197–206.
- Okada, S.; Puel, A.; Casanova, J.L.; Kobayashi, M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin. Transl. Immunol. 2016, 5, e114.
- Puel, A. Human inborn errors of immunity underlying superficial or invasive candidiasis. Hum. Genet. 2020, 139, 1011–1022.
- Millsop, J.W.; Fazel, N. Oral candidiasis. Clin. Dermatol. 2016, 34, 487–494.
- Holland, S.M.; Vinh, D.C. Yeast infections—Human genetics on the rise. N. Engl. J. Med. 2009, 361, 1798–1801.
- Vinh, D.C. Insights into human antifungal immunity from primary immunodeficiencies. Lancet Infect. Dis. 2011, 11, 780–792.
- Lionakis, M.S.; Iliev, I.D.; Hohl, T.M. Immunity against fungi. JCI Insight 2017, 2, e93156.
- Vinh, D.C. The molecular immunology of human susceptibility to fungal diseases: Lessons from single gene defects of immunity. Expert Rev. Clin. Immunol. 2019, 15, 461–486.
- Break, T.J.; Oikonomou, V.; Dutzan, N.; Desai, J.V.; Swidergall, M.; Freiwald, T.; Chauss, D.; Harrison, O.J.; Alejo, J.; Williams, D.W.; et al. Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science 2021, 371, eaay5731.
- Philippot, Q.; Casanova, J.L.; Puel, A. Candidiasis in patients with APS-1: Low IL-17, high IFN-γ, or both? Curr. Opin. Immunol. 2021, 72, 318–323.
- Slatter, M.A.; Gennery, A.R. Advances in the treatment of severe combined immunodeficiency. Clin. Immunol. 2022, 242, 109084.
- Sponzilli, I.; Notarangelo, L.D. Severe combined immunodeficiency (SCID): From molecular basis to clinical management. Acta Biomed. 2011, 82, 5–13.
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 42, 1473–1507.
- Chavoshzadeh, Z.; Darougar, S.; Momen, T.; Esmaeilzadeh, H.; Abolhassani, H.; Cheraghi, T.; van der Burg, M.; van Zelm, M. Immunodeficiencies affecting cellular and humoral immunity. In Inborn Errors of Immunity—A Practical Guide, 1st ed.; Aghamohammadi, A., Abolhassani, H., Rezaei, N., Yazdani, R., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 9–39, Chapter 2.
- Puck, J.M. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia. Immunol. Rev. 2019, 287, 241–252.
- Dorsey, M.J.; Wright, N.A.M.; Chaimowitz, N.S.; Dávila Saldaña, B.J.; Miller, H.; Keller, M.D.; Thakar, M.S.; Shah, A.J.; Abu-Arja, R.; Andolina, J.; et al. Infections in Infants with SCID: Isolation, Infection Screening, and Prophylaxis in PIDTC Centers. J. Clin. Immunol. 2021, 41, 38–50.
- Bergerson, J.R.E.; Freeman, A.F. An Update on Syndromes with a Hyper-IgE Phenotype. Immunol. Allergy Clin. N. Am. 2019, 39, 49–61.
- Olbrich, P.; Freeman, A.F. STAT1 and STAT3 mutations: Important lessons for clinical immunologists. Expert Rev. Clin. Immunol. 2018, 14, 1029–1041.
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776.
- Renner, E.D.; Rylaarsdam, S.; Anover-Sombke, S.; Rack, A.L.; Reichenbach, J.; Carey, J.C.; Zhu, Q.; Jansson, A.F.; Barboza, J.; Schimke, L.F.; et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J. Allergy Clin. Immunol. 2008, 122, 181–187.
- Aggor, F.E.Y.; Break, T.J.; Trevejo-Nuñez, G.; Whibley, N.; Coleman, B.M.; Bailey, R.D.; Kaplan, D.H.; Naglik, J.R.; Shan, W.; Shetty, A.C.; et al. Oral epithelial IL-22/STAT3 signaling licenses IL-17-mediated immunity to oral mucosal candidiasis. Sci. Immunol. 2020, 5, eaba0570.
- Tsilifis, C.; Freeman, A.F.; Gennery, A.R. STAT3 Hyper-IgE Syndrome-an Update and Unanswered Questions. J. Clin. Immunol. 2021, 41, 864–880.
- Yanagimachi, M.; Ohya, T.; Yokosuka, T.; Kajiwara, R.; Tanaka, F.; Goto, H.; Takashima, T.; Morio, T.; Yokota, S. The Potential and Limits of Hematopoietic Stem Cell Transplantation for the Treatment of Autosomal Dominant Hyper-IgE Syndrome. J. Clin. Immunol. 2016, 36, 511–516.
- Harrison, S.C.; Tsilifis, C.; Slatter, M.A.; Nademi, Z.; Worth, A.; Veys, P.; Ponsford, M.J.; Jolles, S.; Al-Herz, W.; Flood, T.; et al. Hematopoietic Stem Cell Transplantation Resolves the Immune Deficit Associated with STAT3-Dominant-Negative Hyper-IgE Syndrome. J. Clin. Immunol. 2021, 41, 934–943.
- Béziat, V.; Li, J.; Lin, J.X.; Ma, C.S.; Li, P.; Bousfiha, A.; Pellier, I.; Zoghi, S.; Baris, S.; Keles, S.; et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci. Immunol. 2018, 3, eaat4956.
- Frey-Jakobs, S.; Hartberger, J.M.; Fliegauf, M.; Bossen, C.; Wehmeyer, M.L.; Neubauer, J.C.; Bulashevska, A.; Proietti, M.; Fröbel, P.; Nöltner, C.; et al. ZNF341 controls STAT3 expression and thereby immunocompetence. Sci. Immunol. 2018, 3, eaat4941.
- Sassi, A.; Lazaroski, S.; Wu, G.; Haslam, S.M.; Fliegauf, M.; Mellouli, F.; Patiroglu, T.; Unal, E.; Ozdemir, M.A.; Jouhadi, Z.; et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J. Allergy Clin. Immunol. 2014, 133, 1410–1419.
- Winslow, A.; Jalazo, E.R.; Evans, A.; Winstead, M.; Moran, T. A De Novo Cause of PGM3 Deficiency Treated with Hematopoietic Stem Cell Transplantation. J. Clin. Immunol. 2022, 42, 691–694.
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018, 378, 1132–1141.
- Constantine, G.M.; Lionakis, M.S. Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol. Rev. 2019, 287, 103–120.
- Bjørklund, G.; Pivin, M.; Hangan, T.; Yurkovskaya, O.; Pivina, L. Autoimmune polyendocrine syndrome type 1: Clinical manifestations, pathogenetic features, and management approach. Autoimmun. Rev. 2022, 21, 103135.
- Goldfarb, Y.; Givony, T.; Kadouri, N.; Dobeš, J.; Peligero-Cruz, C.; Zalayat, I.; Damari, G.; Dassa, B.; Ben-Dor, S.; Gruper, Y.; et al. Mechanistic dissection of dominant AIRE mutations in mouse models reveals AIRE autoregulation. J. Exp. Med. 2021, 218, e20201076.
- Puel, A.; Döffinger, R.; Natividad, A.; Chrabieh, M.; Barcenas-Morales, G.; Picard, C.; Cobat, A.; Ouachée-Chardin, M.; Toulon, A.; Bustamante, J.; et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 2010, 207, 291–297.
- Kisand, K.; Bøe Wolff, A.S.; Podkrajsek, K.T.; Tserel, L.; Link, M.; Kisand, K.V.; Ersvaer, E.; Perheentupa, J.; Erichsen, M.M.; Bratanic, N.; et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 2010, 207, 299–308.
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68.
- Boisson, B.; Wang, C.; Pedergnana, V.; Wu, L.; Cypowyj, S.; Rybojad, M.; Belkadi, A.; Picard, C.; Abel, L.; Fieschi, C.; et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 2013, 39, 676–686.
- Marujo, F.; Pelham, S.J.; Freixo, J.; Cordeiro, A.I.; Martins, C.; Casanova, J.L.; Lei, W.T.; Puel, A.; Neves, J.F. A Novel TRAF3IP2 Mutation Causing Chronic Mucocutaneous Candidiasis. J. Clin. Immunol. 2021, 41, 1376–1379.
- Herjan, T.; Hong, L.; Bubenik, J.; Bulek, K.; Qian, W.; Liu, C.; Li, X.; Chen, X.; Yang, H.; Ouyang, S.; et al. IL-17-receptor-associated adaptor Act1 directly stabilizes mRNAs to mediate IL-17 inflammatory signaling. Nat. Immunol. 2018, 19, 354–365.
- Blanco Lobo, P.; Lei, W.T.; Pelham, S.J.; Guisado Hernández, P.; Villaoslada, I.; de Felipe, B.; Lucena, J.M.; Casanova, J.L.; Olbrich, P.; Puel, A.; et al. Biallelic TRAF3IP2 variants causing chronic mucocutaneous candidiasis in a child harboring a STAT1 variant. Pediatr. Allergy Immunol. 2021, 32, 1804–1812.
- Li, J.; Ritelli, M.; Ma, C.S.; Rao, G.; Habib, T.; Corvilain, E.; Bougarn, S.; Cypowyj, S.; Grodecká, L.; Lévy, R.; et al. Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-β. Sci. Immunol. 2019, 4, eaax7965.
- de Beaucoudrey, L.; Samarina, A.; Bustamante, J.; Cobat, A.; Boisson-Dupuis, S.; Feinberg, J.; Al-Muhsen, S.; Jannière, L.; Rose, Y.; de Suremain, M.; et al. Revisiting human IL-12Rβ1 deficiency: A survey of 141 patients from 30 countries. Medicine 2010, 89, 381–402.
- Prando, C.; Samarina, A.; Bustamante, J.; Boisson-Dupuis, S.; Cobat, A.; Picard, C.; AlSum, Z.; Al-Jumaah, S.; Al-Hajjar, S.; Frayha, H.; et al. Inherited IL-12p40 deficiency: Genetic, immunologic, and clinical features of 49 patients from 30 kindreds. Medicine 2013, 92, 109–122.
- Okada, S.; Markle, J.G.; Deenick, E.K.; Mele, F.; Averbuch, D.; Lagos, M.; Alzahrani, M.; Al-Muhsen, S.; Halwani, R.; Ma, C.S.; et al. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 2015, 349, 606–613.
- Gross, O.; Gewies, A.; Finger, K.; Schäfer, M.; Sparwasser, T.; Peschel, C.; Förster, I.; Ruland, J. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 2006, 442, 651–656.
- LeibundGut-Landmann, S.; Gross, O.; Robinson, M.J.; Osorio, F.; Slack, E.C.; Tsoni, S.V.; Schweighoffer, E.; Tybulewicz, V.; Brown, G.D.; Ruland, J.; et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 2007, 8, 630–638.
- Glocker, E.O.; Hennigs, A.; Nabavi, M.; Schäffer, A.A.; Woellner, C.; Salzer, U.; Pfeifer, D.; Veelken, H.; Warnatz, K.; Tahami, F.; et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009, 361, 1727–1735.
- Gavino, C.; Cotter, A.; Lichtenstein, D.; Lejtenyi, D.; Fortin, C.; Legault, C.; Alirezaie, N.; Majewski, J.; Sheppard, D.C.; Behr, M.A.; et al. CARD9 deficiency and spontaneous central nervous system candidiasis: Complete clinical remission with GM-CSF therapy. Clin. Infect. Dis. 2014, 59, 81–84.
- Gavino, C.; Hamel, N.; Zeng, J.B.; Legault, C.; Guiot, M.C.; Chankowsky, J.; Lejtenyi, D.; Lemire, M.; Alarie, I.; Dufresne, S.; et al. Impaired RASGRF1/ERK-mediated GM-CSF response characterizes CARD9 deficiency in French-Canadians. J. Allergy Clin. Immunol. 2016, 137, 1178–1188.
- Li, J.; Vinh, D.C.; Casanova, J.L.; Puel, A. Inborn errors of immunity underlying fungal diseases in otherwise healthy individuals. Curr. Opin. Microbiol. 2017, 40, 46–57.
- van de Veerdonk, F.L.; Plantinga, T.S.; Hoischen, A.; Smeekens, S.P.; Joosten, L.A.; Gilissen, C.; Arts, P.; Rosentul, D.C.; Carmichael, A.J.; Smits-van der Graaf, C.A.; et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 2011, 365, 54–61.
- Liu, L.; Okada, S.; Kong, X.F.; Kreins, A.Y.; Cypowyj, S.; Abhyankar, A.; Toubiana, J.; Itan, Y.; Audry, M.; Nitschke, P.; et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 2011, 208, 1635–1648.
- Depner, M.; Fuchs, S.; Raabe, J.; Frede, N.; Glocker, C.; Doffinger, R.; Gkrania-Klotsas, E.; Kumararatne, D.; Atkinson, T.P.; Schroeder, H.W., Jr.; et al. The Extended Clinical Phenotype of 26 Patients with Chronic Mucocutaneous Candidiasis due to Gain-of-Function Mutations in STAT1. J. Clin. Immunol. 2016, 36, 73–84.
- Toubiana, J.; Okada, S.; Hiller, J.; Oleastro, M.; Lagos Gomez, M.; Aldave Becerra, J.C.; Ouachée-Chardin, M.; Fouyssac, F.; Girisha, K.M.; Etzioni, A.; et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 2016, 127, 3154–3164.
- Frede, N.; Rojas-Restrepo, J.; Caballero Garcia de Oteyza, A.; Buchta, M.; Hübscher, K.; Gámez-Díaz, L.; Proietti, M.; Saghafi, S.; Chavoshzadeh, Z.; Soler-Palacin, P.; et al. Genetic Analysis of a Cohort of 275 Patients with Hyper-IgE Syndromes and/or Chronic Mucocutaneous Candidiasis. J. Clin. Immunol. 2021, 41, 1804–1838.
- Zheng, J.; van de Veerdonk, F.L.; Crossland, K.L.; Smeekens, S.P.; Chan, C.M.; Al Shehri, T.; Abinun, M.; Gennery, A.R.; Mann, J.; Lendrem, D.W.; et al. Gain-of-function STAT1 mutations impair STAT3 activity in patients with chronic mucocutaneous candidiasis (CMC). Eur. J. Immunol. 2015, 45, 2834–2846.
- Hiller, J.; Hagl, B.; Effner, R.; Puel, A.; Schaller, M.; Mascher, B.; Eyerich, S.; Eyerich, K.; Jansson, A.F.; Ring, J.; et al. STAT1 Gain-of-Function and Dominant Negative STAT3 Mutations Impair IL-17 and IL-22 Immunity Associated with CMC. J. Investig. Dermatol. 2018, 138, 711–714.
- Zimmerman, O.; Olbrich, P.; Freeman, A.F.; Rosen, L.B.; Uzel, G.; Zerbe, C.S.; Rosenzweig, S.D.; Kuehn, H.S.; Holmes, K.L.; Stephany, D.; et al. STAT1 Gain-of-Function Mutations Cause High Total STAT1 Levels With Normal Dephosphorylation. Front. Immunol. 2019, 10, 1433.
- Leiding, J.W.; Okada, S.; Hagin, D.; Abinun, M.; Shcherbina, A.; Balashov, D.N.; Kim, V.H.D.; Ovadia, A.; Guthery, S.L.; Pulsipher, M.; et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J. Allergy Clin. Immunol. 2018, 141, 704–717.e5.
- Kiykim, A.; Charbonnier, L.M.; Akcay, A.; Karakoc-Aydiner, E.; Ozen, A.; Ozturk, G.; Chatila, T.A.; Baris, S. Hematopoietic Stem Cell Transplantation in Patients with Heterozygous STAT1 Gain-of-Function Mutation. J. Clin. Immunol. 2019, 39, 37–44.
- Baris, S.; Alroqi, F.; Kiykim, A.; Karakoc-Aydiner, E.; Ogulur, I.; Ozen, A.; Charbonnier, L.M.; Bakır, M.; Boztug, K.; Chatila, T.A.; et al. Severe Early-Onset Combined Immunodeficiency due to Heterozygous Gain-of-Function Mutations in STAT1. J. Clin. Immunol. 2016, 36, 641–648.
- Weinacht, K.G.; Charbonnier, L.M.; Alroqi, F.; Plant, A.; Qiao, Q.; Wu, H.; Ma, C.; Torgerson, T.R.; Rosenzweig, S.D.; Fleisher, T.A.; et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J. Allergy Clin. Immunol. 2017, 139, 1629–1640.e2.
- Bloomfield, M.; Kanderová, V.; Paračková, Z.; Vrabcová, P.; Svatoň, M.; Froňková, E.; Fejtková, M.; Zachová, R.; Rataj, M.; Zentsová, I.; et al. Utility of Ruxolitinib in a Child with Chronic Mucocutaneous Candidiasis Caused by a Novel STAT1 Gain-of-Function Mutation. J. Clin. Immunol. 2018, 38, 589–601.
- Forbes, L.R.; Vogel, T.P.; Cooper, M.A.; Castro-Wagner, J.; Schussler, E.; Weinacht, K.G.; Plant, A.S.; Su, H.C.; Allenspach, E.J.; Slatter, M.; et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J. Allergy Clin. Immunol. 2018, 142, 1665–1669.
- Borgström, E.W.; Edvinsson, M.; Pérez, L.P.; Norlin, A.C.; Enoksson, S.L.; Hansen, S.; Fasth, A.; Friman, V.; Kämpe, O.; Månsson, R.; et al. Three Adult Cases of STAT1 Gain-of-Function with Chronic Mucocutaneous Candidiasis Treated with JAK Inhibitors. J. Clin. Immunol. 2022.
- Deyà-Martínez, A.; Rivière, J.G.; Roxo-Junior, P.; Ramakers, J.; Bloomfield, M.; Guisado Hernandez, P.; Blanco Lobo, P.; Abu Jamra, S.R.; Esteve-Sole, A.; Kanderova, V.; et al. Impact of JAK Inhibitors in Pediatric Patients with STAT1 Gain of Function (GOF) Mutations-10 Children and Review of the Literature. J. Clin. Immunol. 2022, 42, 1071–1082.
- Immunodeficiencies EESf. Multicentric Retrospective Study on JAKinib Treatment of Patients with IEI of the JAK/STAT Pathway. Available online: https://esid.org/Working-Parties/Inborn-Errors-Working-Party-IEWP/Studies/Multicentric-retrospective-study-on-JAKinib-treatment-of-patients-with-IEI-of-the-JAK-STAT-pathway (accessed on 1 January 2020).
- Ling, Y.; Cypowyj, S.; Aytekin, C.; Galicchio, M.; Camcioglu, Y.; Nepesov, S.; Ikinciogullari, A.; Dogu, F.; Belkadi, A.; Levy, R.; et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med. 2015, 212, 619–631.
- Desai, J.V.; Urban, A.; Swaim, D.Z.; Colton, B.; Kibathi, L.W.; Ferrè, E.M.N.; Stratton, P.; Merideth, M.A.; Hunsberger, S.; Matkovits, T.; et al. Efficacy of Cochleated Amphotericin B in Mouse and Human Mucocutaneous Candidiasis. Antimicrob. Agents Chemother. 2022, 66, e0030822.
- Bruserud, Ø.; Oftedal, B.E.; Landegren, N.; Erichsen, M.M.; Bratland, E.; Lima, K.; Jørgensen, A.P.; Myhre, A.G.; Svartberg, J.; Fougner, K.J.; et al. A Longitudinal Follow-up of Autoimmune Polyendocrine Syndrome Type 1. J. Clin. Endocrinol. Metab. 2016, 101, 2975–2983.
- Koo, S.; Kejariwal, D.; Al-Shehri, T.; Dhar, A.; Lilic, D. Oesophageal candidiasis and squamous cell cancer in patients with gain-of-function STAT1 gene mutation. United Eur. Gastroenterol. J. 2017, 5, 625–631.
- Kullberg, B.J.; Arendrup, M.C. Invasive Candidiasis. N. Engl. J. Med. 2015, 373, 1445–1456.
- Rotulo, G.A.; Plat, G.; Beaupain, B.; Blanche, S.; Moushous, D.; Sicre de Fontbrune, F.; Leblanc, T.; Renard, C.; Barlogis, V.; Vigue, M.G.; et al. Recurrent bacterial infections, but not fungal infections, characterise patients with ELANE-related neutropenia: A French Severe Chronic Neutropenia Registry study. Br. J. Haematol. 2021, 194, 908–920.
- Engel, M.E.; Hickstein, D.D.; Bauer, T.R., Jr.; Calder, C.; Manes, B.; Frangoul, H. Matched unrelated bone marrow transplantation with reduced-intensity conditioning for leukocyte adhesion deficiency. Bone Marrow Transpl. 2006, 37, 717–718.
- Lehrer, R.I.; Cline, M.J. Leukocyte myeloperoxidase deficiency and disseminated candidiasis: The role of myeloperoxidase in resistance to Candida infection. J. Clin. Investig. 1969, 48, 1478–1488.
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B., Jr.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic granulomatous disease. Medicine 2000, 79, 155–169.
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J.; et al. Common severe infections in chronic granulomatous disease. Clin. Infect. Dis. 2015, 60, 1176–1183.
- Kuhns, D.B.; Alvord, W.G.; Heller, T.; Feld, J.J.; Pike, K.M.; Marciano, B.E.; Uzel, G.; DeRavin, S.S.; Priel, D.A.; Soule, B.P.; et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N. Engl. J. Med. 2010, 363, 2600–2610.
- Levy, O.; Bourquin, J.P.; McQueen, A.; Cantor, A.B.; Lachenauer, C.; Malley, R. Fatal disseminated Candida lusitaniae infection in an infant with chronic granulomatous disease. Pediatr. Infect. Dis. J. 2002, 21, 262–264.
- Estrada, B.; Mancao, M.Y.; Polski, J.M.; Figarola, M.S. Candida lusitaniae and chronic granulomatous disease. Pediatr. Infect. Dis. J. 2006, 25, 758–759.
- Corvilain, E.; Casanova, J.L.; Puel, A. Inherited CARD9 Deficiency: Invasive Disease Caused by Ascomycete Fungi in Previously Healthy Children and Adults. J. Clin. Immunol. 2018, 38, 656–693.
- Drummond, R.A.; Lionakis, M.S. Mechanistic Insights into the Role of C-Type Lectin Receptor/CARD9 Signaling in Human Antifungal Immunity. Front. Cell. Infect. Microbiol. 2016, 6, 39.
- Lionakis, M.S. Primary immunodeficiencies and invasive fungal infection: When to suspect and how to diagnose and manage. Curr. Opin. Infect. Dis. 2019, 32, 531–537.
- Drummond, R.A.; Collar, A.L.; Swamydas, M.; Rodriguez, C.A.; Lim, J.K.; Mendez, L.M.; Fink, D.L.; Hsu, A.P.; Zhai, B.; Karauzum, H.; et al. CARD9-Dependent Neutrophil Recruitment Protects against Fungal Invasion of the Central Nervous System. PLoS Pathog. 2015, 11, e1005293.
- Drummond, R.A.; Swamydas, M.; Oikonomou, V.; Zhai, B.; Dambuza, I.M.; Schaefer, B.C.; Bohrer, A.C.; Mayer-Barber, K.D.; Lira, S.A.; Iwakura, Y.; et al. CARD9(+) microglia promote antifungal immunity via IL-1β- and CXCL1-mediated neutrophil recruitment. Nat. Immunol. 2019, 20, 559–570.
- Drewniak, A.; Gazendam, R.P.; Tool, A.T.; van Houdt, M.; Jansen, M.H.; van Hamme, J.L.; van Leeuwen, E.M.; Roos, D.; Scalais, E.; de Beaufort, C.; et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 2013, 121, 2385–2392.
- Goel, S.; Kuehn, H.S.; Chinen, J.; Niemela, J.; Stoddard, J.; Yamanaka, D.; Garofalo, M.; Samir, S.; Migaud, M.; Oikonomou, V.; et al. CARD9 Expression Pattern, Gene Dosage, and Immunodeficiency Phenotype Revisited. J. Clin. Immunol. 2022, 42, 336–349.
- Queiroz-Telles, F.; Mercier, T.; Maertens, J.; Sola, C.B.S.; Bonfim, C.; Lortholary, O.; Constantino-Silva, R.M.N.; Schrijvers, R.; Hagen, F.; Meis, J.F.; et al. Successful Allogenic Stem Cell Transplantation in Patients with Inherited CARD9 Deficiency. J. Clin. Immunol. 2019, 39, 462–469.

