1. Chronic Mucocutaneous Candidiasis (CMC)
Mucocutaneous infections, such as oral thrush or diaper dermatitis, are relatively common in pediatrics. These infections are mostly found in the context of concomitant antibiotic treatment, topical or systemic corticosteroid therapy, or breakdown of the local skin barrier
[1][8]. When mucocutaneous candidiasis is persistent or recurrent and the aforementioned risk factors are absent, the condition is often referred to as “chronic mucocutaneous candidiasis” (CMC). Although this term appears in more than 1000 publications in PubMed, there is no clear definition of this disease state. The original report by Kirkpatrick et al. did not include proposals for the minimal duration or minimal number of recurrences in a defined time period
[2][9]. Of note, some authors use the term “syndromic CMC” in the context of associated autoimmunity, whereas others accept it as an isolated infection-based entity
[2][3][4][9,10,11].
The
C. albicans is the most common isolate causing CMC. When facing a patient with CMC, the physician should review and consider risk factors often associated with this disease, such as: concomitant antibiotic use, topical or systemic corticosteroid therapy, diabetes mellitus, secondary immunosuppression (e.g., chemotherapy and/or radiotherapy for hemato-oncologic diseases), HIV, as well as underlying (congenital) alterations of the immune system
[1][5][8,12].
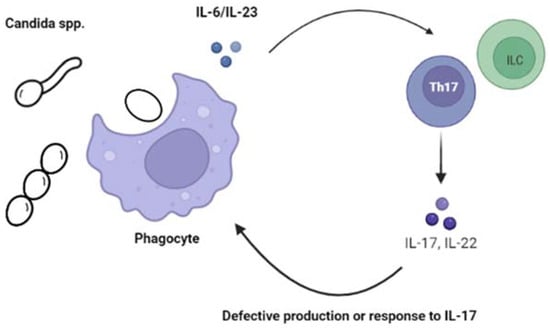
In the last few years, several IEI have been associated with CMC, many of which present in childhood. The study of these rare diseases has provided us with a deeper understanding of the pathophysiologic mechanisms involved. Overall, alterations and imbalances of IL-17 and IL-22, and possibly IFN-γ, have been identified as important factors predisposing individuals to develop CMC
[4][6][7][8][9][10][11][11,13,14,15,16,17,18] Figure 1.
Figure 1.
Alterations of the IL-17 immunity increases the susceptibility to chronic mucocutaneous candidiasis (CMC).
The CMC may be the presenting symptom in patients with inherited T cell deficiencies presenting as severe combined immunodeficiencies ((S)CID). This group is fairly heterogeneous and includes various subtypes that differ in their clinical manifestations and severity, laboratory findings, causal genes, and management
[12][19]. Generally, patients with (S)CID are susceptible to a broad range of infectious agents. Pneumonia from
Pneumocystis jiroveci, another fungal infection, is pathognomonic for T cell deficiencies, including (S)CID, and can be the life-threatening initial presentation
[13][20]. The T cells in (S)CID patients are deficient in numbers and/or function
[14][15][1,21]. In addition, some of these patients may be detected by systematic neonatal screening programs quantifying T cell receptor excision circles (TRECs) in neonatal dried blood spots
[16][22], whereas for others, the diagnosis will be established later, through abnormal lymphocyte subsets and/or immunoglobulin results. The genetic testing for disease-causing mutations in underlying genes will help to definitively establish the molecular diagnosis
[12][19]. The management of these patients differs according to the clinical presentation, the complementary laboratory results, and the affected gene, but often includes infection prophylaxis (isolation, antibiotic and/or antifungal prophylaxis, immunoglobulin replacement), and supportive care (e.g., nutrition)
[17][23]. The early evaluation of curative treatment options such as hematopoietic stem cell transplant (HSCT) or, in selected cases, gene therapy, is necessary
[12][19].
In contrast to patients with (S)CID, other IEI marked by CMC can be associated with discrete syndromes (including the Hyper-IgE syndrome [HIES], or autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy [APECED]); susceptible to other prototypical infections (e.g., S. aureus or mycobacteria); or occur in isolation.
The dominant-negative (DN) STAT3 mutations are responsible for autosomal dominant (AD) HIES, or Job’s syndrome, which is a complex disorder with hematopoietic and non-hematopoietic clinical manifestations. The classical clinical phenotype includes early-onset rash and eczema, bone fractures, delayed dentition, “cold” skin abscesses (due to
S. aureus), recurrent sinopulmonary infections with pneumatocele development, and characteristic facial features
[18][19][24,25]. In addition, the elevated eosinophil counts and IgE levels in blood are characteristic laboratory features for HIES, but they are not always present, or may be present intermittently. The CMC is a key manifestation of HIES due to DN-STAT3 mutations. Further, the invasive
Candida infection is rare, but patients with previous lung damage are at risk of invasive aspergillosis (see section below). Impaired Th17 differentiation with decreased proportions of IL-17- and IL-22-producing T cells is likely responsible for the increased susceptibility to mucocutaneous
Candida and
S aureus infections in DN-STAT3 HIES
[20][21][22][26,27,28]. The management of this IEI consists of antibiotic prophylaxis, supportive care, and antifungal prophylaxis when lung damage has occurred. The early treatment of potential infections is recommended, as many patients may have important bacterial infections without displaying significant inflammatory signs
[18][23][24,29]. Further, hematopoietic stem cell transplantation has been performed to date in a small group of patients, restoring some immunologic alterations. However, non-hematological complications such as vasculopathy or bone-related complications will most likely not benefit from this procedure
[23][24][25][29,30,31].
The CMC may be seen in other causes of HIES. For example, patients with autosomal recessive (AR) mutations in
ZNF341 [26][27][32,33] and
PGM3 [28][29][34,35] show disease manifestations resembling DN-STAT3 HIES, including CMC. The ZNF341 is a transcription factor that binds to the STAT3 promoter. In addition, the biallelic mutations in
ZNF341 lead to the loss of its function, decreasing STAT3 production and, thus, its function. Further, the ZNF341 deficiency is managed similarly to DN-STAT3 HIES. PGM3 is a congenital disorder of glycosylation and has been occasionally reported in association with CMC
[28][34]. The exact mechanism by which biallelic mutations in PGM3 increase susceptibility to CMC is not clear, although slightly decreased Th17 levels have been described
[28][34].
The CMC is one of the most common presenting symptoms in children with autoimmune polyglandular syndrome type 1 (APS-1, also named APECED; OMIM 240300): 25–50% of affected patients present with the CMC in the first year of life, with rates reaching 80–90% in the adult population
[30][31][32][36,37,38]. The APECED is a rare (1:100,000) monogenic IEI due to mutations in
AIRE, that are classically autosomal-recessive (AR), although dominant-negative variants have also been reported
[33][39]. The typical APECED patients present with CMC, hypoparathyroidism, and primary adrenal insufficiency, although other autoimmune manifestations, such as pneumonitis and enteropathy, as well as enamel hypoplasia, have been described. In APECED, loss of AIRE function results in thymic dysfunction with the escape of autoreactive T cells. The lymphocytic organ infiltration, in combination with the generation of anti-cytokine autoantibodies, causes the most characteristic disease manifestations. The autoantibodies to IL-17 have been traditionally associated with CMC
[34][35][40,41]. However, recent data focusing on gingival tissue suggest a more complex interaction beyond circulating T cells and include impaired type 17 mucosal immunity as well as immunopathology promoted by excessive type 1 mucosal inflammation
[10][11][17,18]. The contribution of the latter mechanism may be supported by the therapeutic effect of targeted treatment strategies such as JAK inhibitors (JAKinibs)
[10][17].
The CMC may also be due to IEI without one of the above syndromes, but in association with increased susceptibility to other infections. However, the DN-STAT3 HIES demonstrated the potential non-redundancy of the IL-17 pathway in susceptibility to CMC and
S. aureus skin and soft tissue infection (SSTI). The discovery of AD IL-17F and AR IL-17RA deficiencies underlying CMC pinpointed the critical nature of this cytokine in human immunity to
Candida [36][42]. In addition to CMC, patients may also develop staphylococcal SSTI. Consistent with this pathophysiological paradigm, AR ACT1 deficiency is another IEI that can manifest with CMC and
S. aureus SSTI
[37][38][43,44]. ACT1 is an adapter protein recruited to the IL-17 receptor, where it binds the IL17RA subunit, and mediates downstream signaling
[39][45]. The biallelic mutations in the gene encoding ACT1,
TRAF3IP2, result in impaired NF-κB activation. In a case series of published reports (n = 12)
[40][46], CMC occurred in early childhood (before age 2 years) in 80% of cases, while
S. aureus SSTI were documented in about 50% of cases. The standard immune phenotyping and immunoglobulin levels were unremarkable in most patients. However, the treatment responses of CMC were satisfactory when documented, and no fatal cases have been described
[40][46]. The AD JNK1 deficiency has only been reported in three individuals from one family
[41][47]. The JNK1 protein is part of the IL-17 and TGFβ1 signaling pathways. TGFβ1 is involved in the Th17 differentiation process, and its compromise due to mutant JNK1 likely explains the reduced proportion of ex vivo and in vitro differentiated Th17 cells found in all patients. Similar to the aforementioned etiologies, the patients’ clinical phenotype involved early-onset CMC and
S. aureus SSTI. In addition, all patients had features suggestive of an Ehlers-Danlos such as connective tissue disorder, most likely due to the abnormal TGFβ1 signaling
[41][47].
The susceptibility to CMC can also occur in association with ‘intracellular pathogens,’ notably mycobacteria. In patients with autosomal recessive IL-12p40 and IL-12RB1 deficiency, CMC can occur in ~25% of patients, although it is not necessarily concomitant with other infections
[42][43][48,49]. Similarly, patients with autosomal recessive RORγt deficiency, due to biallelic mutations in
RORC (encoding the RORγt transcription factor fundamental for regulating Th17 development), have susceptibility to both CMC as well as non-tuberculous mycobacterial infections
[44][50]. These IEI can also predispose to invasive disease with other fungi (see below).
The CARD9 deficiency is the only known IEI that predisposes to both CMC and invasive candidiasis. The CARD9 encodes an adaptor protein associated with multiple C-type lectin receptors (CLRs), such as Dectin-1, which are involved in the recognition of fungus and subsequent pro-inflammatory response
[45][46][51,52]. The loss of CARD9 function leads to variably diminished, but not abrogated, Th17 responses, potentially contributing to occasional CMC
[47][48][49][50][53,54,55,56]. More strikingly, CARD9 deficiency leads to spontaneous development of invasive candidiasis and, distinctly, to central nervous system (CNS) involvement
(see the Section 2.1.2 below).
The most common IEI in the context of CMC are STAT1 gain of function (GOF) mutations
[51][52][53][57,58,59]. In some CMC cohorts, about half of the cases were diagnosed with this disease
[51][54][57,60]. In a large international cohort of patients with such features, the search for underlying gene defects using a targeted sequencing approach yielded a diagnosis in 37.5% (24/64) of those with CMC, including: STAT1 GOF (63%), CARD9 (17%), STAT3 (8%), IL17RA (8%), and AIRE (4%)
[55][61]. Of note, these results were obtained from a cohort that included many patients from Middle Eastern countries. Consanguinity is more common and may favor AR disorders, whereas cohorts with patients of European ancestry would likely reveal higher proportions of STAT1 and STAT3 defects
[19][55][25,61].
Although the precise pathophysiological mechanisms by which STAT1 GOF predisposes to CMC still need to be elucidated, it appears that at least one determinant of disease is that increased activation of the JAK-STAT1 pathway results in unbalanced Th17 differentiation
[56][57][58][62,63,64]. The patients with an autosomal dominant (AD) STAT1 GOF mutation present most commonly with early onset (first 2 years of life) CMC. Other disease manifestations, such as recurrent (myco-) bacterial, viral, and non-
Candida fungal infections, have also been reported. Although patients may show reduced numbers of T cells and hypogammaglobulinemia, it is also not uncommon that standard immunologic evaluations with lymphocyte subsets (including Th17), immunoglobulin levels, and vaccine responses are normal. Therefore, genetic testing should be pursued early in such an evaluation. The significant (multiorgan) autoimmune manifestations, vascular abnormalities (aneurysms), and an increased risk of malignancies (squamous cancer) are also part of the broad clinical phenotype. The management of these patients is challenging as it often requires the combination of immunosuppression as well as anti-infective therapy
[18][54][24,60]. In addition, hematopoietic stem cell transplantation (HSCT) is the only curative treatment option, but the current literature indicates high rates of secondary graft failure and mortality
[59][60][65,66]. The JAKinibs such as ruxolitinib or baricitinib, have been shown to effectively treat many of the aforementioned disease manifestations, and in particular CMC
[61][62][63][64][65][67,68,69,70,71]. A recent study summarized the experience with JAKinibs in pediatric STAT1 GOF patients and showed a good response rate (82%; 18/22 subjects) after 1–8 weeks of therapy. Further, most patients were able to discontinue previously prescribed antifungal prophylaxis
[66][72]. It is noteworthy that, although JAK inhibition has now been used in a considerable number of patients, no guidelines exist regarding optimal dosing, monitoring, or follow-up. However, the long-term effects of JAK inhibition in STAT1 GOF, especially in the pediatric population, are yet unknown. Recently, a multinational consortium under the umbrella of ESID/IEWP and ERN has started to elaborate a consensus guideline aiming to address the aforementioned uncertainties
[67][73].
In distinction to the above IEI, isolated CMC (to date) has been described in 3 subjects from different families with AR-complete IL-17RC deficiency
[68][74]. Whether this IEI represents a finite CMC susceptibility, a phenotype in progress that will be revealed with time, or an additional reported case, it cannot be addressed currently.
The CMC management system is not standardized. Overall, acute and infrequently recurring (<2x/year) OPC episodes can likely be successfully treated with oral fluconazole for 3–4 weeks if congruent with antifungal susceptibility testing of isolates. In the case of azole resistance, echinocandins may be an appropriate alternative, although they currently require intravenous administration. Patients with frequently recurring (>3x/year) or persistent CMC should receive secondary prophylaxis with either triazoles or an oral cochleated amphotericin solution, which shows promise and may become an alternative option
[69][75]. It should be noted that inadequately treated CMC may have important sequelae: For both APECED and STAT1 GOF, CMC most commonly affects the oral and esophageal mucosa and may lead to esophageal strictures and stenosis, while in some cases, squamous cell cancer has been reported as a long-term complication
[30][54][70][71][36,60,76,77].
2. Invasive Candidiasis (IC)
The IC is a growing health care problem and is considered the most common fungal disease among hospitalized patients in the developed world
[72][3]. Candidemia, chronic disseminated candidiasis (previously known as hepatosplenic candidiasis), and CNS disease (e.g., meningitis) are often life-threatening and associated with important sequelae. Early diagnosis and prevention are key to avoiding deleterious complications. Risk factors in children include prematurity, damage to the gastrointestinal epithelial or skin (e.g., surgery, indwelling catheter, chemotherapy-associated mucositis, alteration of the microbiota due to the use of broad-spectrum antibiotics), as well as pharmacologic immunosuppression (e.g., corticosteroids or chemotherapy), and of particular interest, a limited number of IEI
[1][50][8,56].
In a patient with IC but lacking the above-mentioned iatrogenic risk factors, an underlying IEI should be considered and investigated. Specifically, IEI with alterations in the number or function of phagocytes should be ruled out. In this regard, IC has been reported in patients with congenital neutropenia syndromes (ELANE, HAX1, etc.)
[73][78] and leukocyte adhesion disorders 1 (LAD-1, ITKB2)
[74][79]. Similarly, complete myeloperoxidase (MPO) deficiency or chronic granulomatous disease (CGD) have been associated with deep-seated
Candida infections
[75][76][77][80,81,82]. The defective production of reactive oxygen species, which is required for an effective oxidative burst permitting elimination of
Candida and other stereotypical microorganisms (e.g.,
Aspergillus; specific bacteria), is the most likely responsible pathophysiologic mechanism
[77][78][82,83]. Of note, the rates of
C. lusitaniae, a fairly uncommon
Candida species, are substantially higher in CGD patients, potentially indicating a specific relevance of oxidative burst for this pathogen
[79][80][84,85].
As stated above, patients with AR CARD9 deficiency are at risk for superficial and, more strikingly, invasive candidiasis (as well as other fungal infections). Importantly, these infections can manifest at any age
[9][50][81][82][16,56,86,87]. The IC may affect various organs such as the bones, the gastrointestinal tract, and the eyes. CNS candidiasis (or meningoencephalitis) is, however, the most characteristic disease manifestation for patients with CARD9 deficiency. Therefore, children as well as adults presenting with spontaneous CNS candidiasis (e.g., meningitis, abscess), in the absence of obvious risk factors such as intraventricular shunts or head surgery, should be evaluated for this IEI
[7][50][81][83][14,56,86,88]. The reason for this increased predilection of
Candida infections to the CNS in patients with CARD9 deficiency has not been elucidated yet. However, intriguingly, CNS candidiasis is often associated with a mitigated neutrophilic response (tissue neutropenia). Candidalysin (a cytolytic peptide toxin produced by various
Candida spp.) induces local microglia to produce interleukin IL1β and the C-X-C motif chemokine in a CARD9-dependent manner, enabling the recruitment of neutrophils to the CNS
[84][85][89,90]. In addition, CARD9-deficient neutrophils have a diminished capacity to kill unopsonized yeast
[86][91]. Although a number of patients have been described as having eosinophilia, raised IgE levels, or even a CVID-like phenotype
[87][92], normal results in immunologic evaluations do not exclude this diagnosis, and thus genetic studies are necessary to establish a definitive diagnosis. Treatment is challenging and consists of intensive and prolonged (sometimes life-long) antifungal therapy. HSCT has been successfully performed in some patients
[88][93].