 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | José Pérez de la Lastra | -- | 2953 | 2023-02-01 10:36:51 | | | |
| 2 | Jason Zhu | Meta information modification | 2953 | 2023-02-02 03:13:20 | | |
Video Upload Options
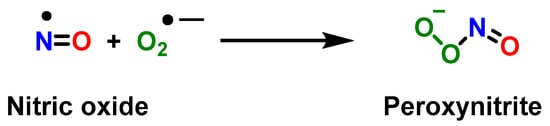
Myeloid-derived suppressor cells MDSCs are a heterogeneous population of cells that expand beyond their physiological regulation during pathologies such as cancer, inflammation, bacterial, and viral infections. Their key feature is their remarkable ability to suppress T cell and natural killer NK cell responses. Certain risk factors for severe COVID-19 disease, such as obesity and diabetes, are associated with oxidative stress. The resulting inflammation and oxidative stress can negatively impact the host. Similarly, cancer cells exhibit a sustained increase in intrinsic ROS generation that maintains the oncogenic phenotype and drives tumor progression. By disrupting endoplasmic reticulum calcium channels, intracellular ROS accumulation can disrupt protein folding and ultimately lead to proteostasis failure. In cancer and COVID-19, MDSCs consist of the same two subtypes (PMN-MSDC and M-MDSC). While the main role of polymorphonuclear MDSCs is to dampen the response of T cells and NK killer cells, they also produce reactive oxygen species ROS and reactive nitrogen species RNS.
1. Introduction
2. Myeloid-Derived Suppressor Cells MDSC and Its Role in Immune System
3. Role of Myeloid-Derived Suppressor Cells in Cancer
4. Role of Myeloid-Derived Suppressor Cells in COVID-19
References
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701.
- De Kleer, I.; Willems, F.; Lambrecht, B.; Goriely, S. Ontogeny of myeloid cells. Front. Immunol. 2014, 5, 423.
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752.
- Millrud, C.R.; Bergenfelz, C.; Leandersson, K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017, 8, 3649.
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835.
- Morales, J.K.; Kmieciak, M.; Knutson, K.L.; Bear, H.D.; Manjili, M.H. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1-bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res. Treat. 2010, 123, 39–49.
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T. INAUGURAL ARTICLE by a Recently Elected Academy Member: Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+ Ly6C+ granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248.
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174.
- Serafini, P.; Bronte, V. Myeloid-derived suppressor cells in tumor-induced T cell suppression and tolerance. In Tumor-Induced Immune Suppression; Springer: Berlin/Heidelberg, Germany, 2014; pp. 99–150.
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+ CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421.
- Cassetta, L.; Bruderek, K.; Skrzeczynska-Moncznik, J.; Osiecka, O.; Hu, X.; Rundgren, I.M.; Lin, A.; Santegoets, K.; Horzum, U.; Godinho-Santos, A. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J. Immunother. Cancer 2020, 8, e001223.
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-derived suppressor cells: Immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol. 2018, 200, 422–431.
- Gimeno, R.; Barquinero, J. Myeloid-derived suppressor cells (MDSC): Another player in the orchestra. Inmunología 2011, 30, 45–53.
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110.
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498.
- Lang, S.; Bruderek, K.; Kaspar, C.; Höing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell SubsetsCD16+/CD11b+ PMN-MDSC in Head and Neck Cancer. Clin. Cancer Res. 2018, 24, 4834–4844.
- Sarkar, T.; Dhar, S.; Sa, G. Tumor-infiltrating T-regulatory cells adapt to altered metabolism to promote tumor-immune escape. Curr. Res. Immunol. 2021, 2, 132–141.
- Burke, M.; Choksawangkarn, W.; Edwards, N.; Ostrand-Rosenberg, S.; Fenselau, C. Exosomes from myeloid-derived suppressor cells carry biologically active proteins. J. Proteome Res. 2014, 13, 836–843.
- Bronte, V.; Chappell, D.B.; Apolloni, E.; Cabrelle, A.; Wang, M.; Hwu, P.; Restifo, N.P. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J. Immunol. 1999, 162, 5728–5737.
- Waight, J.D.; Hu, Q.; Miller, A.; Liu, S.; Abrams, S.I. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS ONE 2011, 6, e27690.
- Raber, P.; Ochoa, A.C.; Rodríguez, P.C. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: Mechanisms of T cell suppression and therapeutic perspectives. Immunol. Investig. 2012, 41, 614–634.
- Koehn, B.H.; Apostolova, P.; Haverkamp, J.M.; Miller, J.S.; McCullar, V.; Tolar, J.; Munn, D.H.; Murphy, W.J.; Brickey, W.J.; Serody, J.S.; et al. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood 2015, 126, 1621–1628.
- Marigo, I.; Bosio, E.; Solito, S.; Mesa, C.; Fernandez, A.; Dolcetti, L.; Ugel, S.; Sonda, N.; Bicciato, S.; Falisi, E.; et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010, 32, 790–802.
- Ku, A.W.; Muhitch, J.B.; Powers, C.A.; Diehl, M.; Kim, M.; Fisher, D.T.; Sharda, A.P.; Clements, V.K.; O’Loughlin, K.; Minderman, H. Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymph nodes. Elife 2016, 5, e17375.
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807.
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of Tumor-Derived Prostaglandin-E2 Blocks the Induction of Myeloid-Derived Suppressor Cells and Recovers Natural Killer Cell ActivityRescue of NK Cells by Blocking the Induction of MDSCs. Clin. Cancer Res. 2014, 20, 4096–4106.
- Tsukamoto, H.; Nishikata, R.; Senju, S.; Nishimura, Y. Myeloid-Derived Suppressor Cells Attenuate TH1 Development through IL-6 Production to Promote Tumor ProgressionDampening of Antitumor TH1 Development by MDSC-Derived IL-6. Cancer Immunol. Res. 2013, 1, 64–76.
- Pan, P.-Y.; Ma, G.; Weber, K.J.; Ozao-Choy, J.; Wang, G.; Yin, B.; Divino, C.M.; Chen, S.-H. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010, 70, 99–108.
- Kwak, T.; Wang, F.; Deng, H.; Condamine, T.; Kumar, V.; Perego, M.; Kossenkov, A.; Montaner, L.J.; Xu, X.; Xu, W. Distinct populations of immune-suppressive macrophages differentiate from monocytic myeloid-derived suppressor cells in cancer. Cell Rep. 2020, 33, 108571.
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453.
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51.
- Su, Y.-L.; Banerjee, S.; White, S.V.; Kortylewski, M. STAT3 in tumor-associated myeloid cells: Multitasking to disrupt immunity. Int. J. Mol. Sci. 2018, 19, 1803.
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185.
- Fultang, N.; Li, X.; Li, T.; Chen, Y.H. Myeloid-derived suppressor cell differentiation in cancer: Transcriptional regulators and enhanceosome-mediated mechanisms. Front. Immunol. 2021, 11, 619253.
- Waight, J.D.; Netherby, C.; Hensen, M.L.; Miller, A.; Hu, Q.; Liu, S.; Bogner, P.N.; Farren, M.R.; Lee, K.P.; Liu, K. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J. Clin. Investig. 2013, 123, 4464–4478.
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962.
- Lapurga, G.; Sun, S.; Carlson, E.; Savardekar, H.; Kendra, K.; Peterson, B.; Carson, W. 686 Characterization of a novel compound that inhibits peroxynitrite generation by myeloid derived suppressor cells. J. Immunother. Cancer 2021, 9, A714.
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802.
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244.
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150.
- Park, S.M.; Youn, J.I. Role of myeloid-derived suppressor cells in immune checkpoint inhibitor therapy in cancer. Arch. Pharmacal Res. 2019, 42, 560–566.
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra67.
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; Andreu-Agullo, C.; Derbyshire, M.L.; Posada, J.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840.e18.
- Horikawa, N.; Abiko, K.; Matsumura, N.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Murakami, R.; Taki, M.; Ukita, M.; Hosoe, Y.; et al. Anti-VEGF therapy resistance in ovarian cancer is caused by GM-CSF-induced myeloid-derived suppressor cell recruitment. Br. J. Cancer 2020, 122, 778–788.
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198.
- Mantovani, A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur. J. Immunol. 2010, 40, 3317–3320.
- Yang, Z.Z.; Ansell, S.M. The role of Treg cells in the cancer immunological response. Am. J. Immunol. 2009, 5, 17–28.
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36.
- Tsuchiya, H.; Shiota, G. Immune evasion by cancer stem cells. Regen. Ther. 2021, 17, 20–33.
- Gabrilovich, D.I.; Bronte, V.; Chen, S.-H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425.
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59.
- Koushki, K.; Salemi, M.; Miri, S.M.; Arjeini, Y.; Keshavarz, M.; Ghaemi, A. Role of myeloid-derived suppressor cells in viral respiratory infections; hints for discovering therapeutic targets for COVID-19. Biomed. Pharmacother. 2021, 144, 112346.
- Fullerton, J.N.; O’Brien, A.J.; Gilroy, D.W. Pathways mediating resolution of inflammation: When enough is too much. J. Pathol. 2013, 231, 8–20.
- Chen, I.-Y.; Moriyama, M.; Chang, M.-F.; Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019, 10, 50.
- Yamada, T.; Sato, S.; Sotoyama, Y.; Orba, Y.; Sawa, H.; Yamauchi, H.; Sasaki, M.; Takaoka, A. RIG-I triggers a signaling-abortive anti-SARS-CoV-2 defense in human lung cells. Nat. Immunol. 2021, 22, 820–828.
- Yin, X.; Riva, L.; Pu, Y.; Martin-Sancho, L.; Kanamune, J.; Yamamoto, Y.; Sakai, K.; Gotoh, S.; Miorin, L.; De Jesus, P.D. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep. 2021, 34, 108628.
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 992–994.
- Scozzi, D.; Cano, M.; Ma, L.; Zhou, D.; Zhu, J.H.; O’Halloran, J.A.; Goss, C.; Rauseo, A.M.; Liu, Z.; Sahu, S.K. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 2021, 6, e143299.
- Bime, C.; Casanova, N.G.; Nikolich-Zugich, J.; Knox, K.S.; Camp, S.M.; Garcia, J.G. Strategies to DAMPen COVID-19-mediated lung and systemic inflammation and vascular injury. Transl. Res. 2021, 232, 37–48.
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353.
- Land, W.G. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome—With a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. 2021, 22, 141–160.
- Parthasarathy, U.; Martinelli, R.; Vollmann, E.H.; Best, K.; Therien, A.G. The impact of DAMP-mediated inflammation in severe COVID-19 and related disorders. Biochem. Pharmacol. 2022, 195, 114847.
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999.
- Grasselli, G.; Tonetti, T.; Protti, A.; Langer, T.; Girardis, M.; Bellani, G.; Laffey, J.; Carrafiello, G.; Carsana, L.; Rizzuto, C. Pathophysiology of COVID-19-associated acute respiratory distress syndrome: A multicentre prospective observational study. Lancet Respir. Med. 2020, 8, 1201–1208.
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859.
- Moderbacher, C.R.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell 2020, 183, 996–1012.e19.
- Shah, V.K.; Firmal, P.; Alam, A.; Ganguly, D.; Chattopadhyay, S. Overview of immune response during SARS-CoV-2 infection: Lessons from the past. Front. Immunol. 2020, 11, 1949.
- Anand, U.; Jakhmola, S.; Indari, O.; Jha, H.C.; Chen, Z.-S.; Tripathi, V.; de la Lastra, J.M.P. Potential therapeutic targets and vaccine development for SARS-CoV-2/COVID-19 pandemic management: A review on the recent update. Front. Immunol. 2021, 12, 658519.
- Gold, M.S.; Sehayek, D.; Gabrielli, S.; Zhang, X.; McCusker, C.; Ben-Shoshan, M. COVID-19 and comorbidities: A systematic review and meta-analysis. Postgrad. Med. 2020, 132, 749–755.
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, immunity, and COVID-19: How age influences the host immune response to coronavirus infections? Front. Physiol. 2021, 11, 571416.
- Latif, M.B.; Shukla, S.; Estrada, P.M.d.R.; Ribeiro, S.P.; Sekaly, R.P.; Sharma, A.A. Immune mechanisms in cancer patients that lead to poor outcomes of SARS-CoV-2 infection. Transl. Res. 2022, 241, 83–95.
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362.
- Tang, G.; Huang, M.; Luo, Y.; Liu, W.; Lin, Q.; Mao, L.; Wu, S.; Xiong, Z.; Hou, H.; Sun, Z. The dynamic immunological parameter landscape in coronavirus disease 2019 patients with different outcomes. Front. Immunol. 2021, 12, 697622.
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 2020, 181, 1489–1501.
- Zhao, J.; Yuan, Q.; Wang, H.; Liu, W.; Liao, X.; Su, Y.; Wang, X.; Yuan, J.; Li, T.; Li, J. Antibody responses to SARS-CoV-2 in patients with novel coronavirus disease 2019. Clin. Infect. Dis. 2020, 71, 2027–2034.
- Suthar, M.S.; Zimmerman, M.G.; Kauffman, R.C.; Mantus, G.; Linderman, S.L.; Hudson, W.H.; Vanderheiden, A.; Nyhoff, L.; Davis, C.W.; Adekunle, O. Rapid generation of neutralizing antibody responses in COVID-19 patients. Cell Rep. Med. 2020, 1, 100040.
- Özkan, B.; Lim, H.; Park, S.-G. Immunomodulatory function of myeloid-derived suppressor cells during B cell-mediated immune responses. Int. J. Mol. Sci. 2018, 19, 1468.
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: Roles in tumor progression. Front. Immunol. 2019, 10, 771.
- Dean, M.J.; Ochoa, J.B.; Sanchez-Pino, M.; Zabaleta, J.; Garai, J.; Del Valle, L.; Wyczechowska, D.; Buckner, L.; Philbrook, P.; Majumder, R. Transcriptome and functions of granulocytic myeloid-derived suppressor cells determine their association with disease severity of COVID-19. medRxiv 2021.
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573.
- Pyzer, A.R.; Cole, L.; Rosenblatt, J.; Avigan, D.E. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int. J. Cancer 2016, 139, 1915–1926.
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790.
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642.
- Ohl, K.; Tenbrock, K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front. Immunol. 2018, 9, 2499.
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835.
- Sacchi, A.; Grassi, G.; Bordoni, V.; Lorenzini, P.; Cimini, E.; Casetti, R.; Tartaglia, E.; Marchioni, L.; Petrosillo, N.; Palmieri, F. Early expansion of myeloid-derived suppressor cells inhibits SARS-CoV-2 specific T-cell response and may predict fatal COVID-19 outcome. Cell Death Dis. 2020, 11, 921.
- Reizine, F.; Lesouhaitier, M.; Gregoire, M.; Pinceaux, K.; Gacouin, A.; Maamar, A.; Painvin, B.; Camus, C.; Le Tulzo, Y.; Tattevin, P. SARS-CoV-2-induced ARDS associates with MDSC expansion, lymphocyte dysfunction, and arginine shortage. J. Clin. Immunol. 2021, 41, 515–525.

