Myeloid-derived suppressor cells MDSCs are a heterogeneous population of cells that expand beyond their physiological regulation during pathologies such as cancer, inflammation, bacterial, and viral infections. Their key feature is their remarkable ability to suppress T cell and natural killer NK cell responses. Certain risk factors for severe COVID-19 disease, such as obesity and diabetes, are associated with oxidative stress. The resulting inflammation and oxidative stress can negatively impact the host. Similarly, cancer cells exhibit a sustained increase in intrinsic ROS generation that maintains the oncogenic phenotype and drives tumor progression. By disrupting endoplasmic reticulum calcium channels, intracellular ROS accumulation can disrupt protein folding and ultimately lead to proteostasis failure. In cancer and COVID-19, MDSCs consist of the same two subtypes (PMN-MSDC and M-MDSC). While the main role of polymorphonuclear MDSCs is to dampen the response of T cells and NK killer cells, they also produce reactive oxygen species ROS and reactive nitrogen species RNS.
- myeloid-derived suppressor cells
- cancer
- COVID-19
1. Introduction
2. Myeloid-Derived Suppressor Cells MDSC and Its Role in Immune System
Under physiological conditions, myeloid progenitor cells differentiate into macrophages M, dendritic cells DC, or granulocytes G [2]. MDSCs are formed from bone marrow precursors when myelopoietic processes are disrupted, which occur when various diseases are triggered [3]. Under certain pathological conditions, such as in cancer or infection, myelopoiesis (defined as the production of the bone marrow and the resulting cells: eosinophilic granulocytes, basophilic granulocytes, neutrophilic granulocytes, and monocytes) is abnormal, allowing the accumulation and proliferation of immature myeloid cells that have potent immunosuppressive capabilities [4,5,6,7][4][5][6][7]. MDSCs were described more than 30 years ago in cancer patients [8]. Common features of MDSCs are their myeloid origin, their immature state and a remarkable ability to suppress T-cell and NK-cell responses [9]. MDSCs are elevated in virtually all patients with cancer and malignancies, and include two main subpopulations of cells: monocytic M-MDSC and granulocytic (polymorphonuclear PMN-MDSC), defined by their expression of plasma membrane markers and their content of immunosuppressive molecules [10]. MDSCs are pathologically activated neutrophils and monocytes and have potent immunosuppressive activity, regulating immune responses in many pathological conditions (including cancer, chronic infection, sepsis, and autoimmunity) and are closely associated with poor prognosis in cancer. MDSCs are a major obstacle to immunotherapies, as accumulation of MDSC populations in circulating leukocytes and tumor infiltrates has been observed in patients who do not respond to checkpoint inhibitor therapy [11,12][11][12]. In addition to their suppressive effects on adaptive immune responses, MDSCs regulate innate immune responses by modulating macrophage cytokine production [13]. Non-immunological functions of MDSCs, such as the promotion of tumor angiogenesis and metastasis, have also been described [14]. In pregnancy and neonates, the functions of MDSCs have been described under physiological conditions [15]. MDSCs are multifaceted and use multiple mechanisms to inhibit both adaptive and innate immunity; for example, in the adaptive system, T cells are a primary target. Initial studies showed that MDSCs produce some of their suppressive effects by releasing soluble mediators [16], requiring cell contact due to the short half-life and distribution of the effector molecules. On the one hand, infiltrating T cells are reduced in the tumor microenvironment [17], and at the same time MDSCs limit the migration of T cells to lymph nodes where they could be activated. Recent studies have shown that suppressive activity is also mediated by MDSC-derived exosomes [18]. Activation of MDSCs is mediated by the expression of inflammatory cytokines, such as GM-CSF, IL-6, G-CSF, IL-1β, PGE2, TNF-α, and VEGF and by transcriptional regulators including STAT3, CEBP/β, STAT5, IRF8, S100A8/9, RB, TIPE2, and GCN2 [4,5,6,19][4][5][6][19]. Growing tumors produce cytokines and other substances that affect the development of MDSC, such as colony-stimulating factors G-CSF and GM-CSF and the MDSC-promoting interleukin IL-6 [20]. Recent results in both tumor mice and cancer patients suggest that increased metabolism of ARG1 by MDSCs inhibits T-cell responses [21]. MDSCs prevent T-cell activation by limiting the availability of amino acids necessary for T-cell proliferation, such as arginine, or by producing substances that block antigen recognition. MDSCs produce arginase 1 ARG1, which competes for the substrate arginine, depleting it, resulting in the loss of the T-cell receptor chain essential for T-cell activation. The main targets of MDSCs are T cells and the main factors involved in immune suppression include ARG1 arginase, iNOS, TGF-β, IL-10, COX2, cysteine sequestration by indoleamine 2,3-dioxygenase IDO, decreased L-selectin expression by T cells, and several others. M-MDSCs and PMN-MDSCs use different immune suppression mechanisms, the former M-type suppress T-cell responses both specifically and non-specifically using mechanisms associated with •NO and cytokine production [22]. PMN-MDSCs can suppress immune responses primarily in an antigen-specific manner and ROS production is essential to maintain this ability [23]. Extravasation of T cells from the blood and lymphatics to the lymph nodes requires the expression of L-selectin/CD62L on T cells. MDSCs express the enzyme ADAM-17, which cleaves L-selectin on T cells, thus preventing extravasation and limiting T-cell entry into lymph nodes [24]. NK-cell cytotoxicity is also inhibited by MDSC [25]. A novel subset of MDSCs specifically targeting NK cells is accumulated in the tumor microenvironment of mice by the proinflammatory cytokine IL-1. Upon their activation by prostaglandin E2 PGE2, MDSCs reduce NK-cell activity in melanoma patients by producing the immunosuppressive transforming growth factor, TGF-1β [26]. Dendritic cells DC are negatively affected by MDSC in a similar way [27], by the suppression of antigen presentation by type 1-T helper cells Th1 [28]. IL-10 and interferon IFN-γ are required for the development of T regulatory cells Tregs, where ARG1 and CD40 play a role in this process. MDSC can alter the production of cytokines. Mice with tumors have decreased IL-7 and STAT5 signaling, which is important for B-cell differentiation, resulting in decreased circulating IgG levels. The population of tumor-associated macrophages (TAMs) promotes tumor progression. M-MDSC-derived macrophages retained most of the properties of their predecessors, including immunosuppressive function [29]. In hypoxic regions of solid tumors, M-MDSCs rapidly convert to TAMs and MDSCs also communicate with macrophages to enhance the protumoral activity of TAMs [30]. STAT3 is a repressor of anti-tumor immunity, and its expression impairs antigen presentation and inhibits the production of immunostimulatory cytokines, while promoting the expression of immunosuppressive molecules. This factor is present in most cancers and induces the production of inflammatory cytokines and growth factors such as IL-6, IL-10, IL-23, LIF, VEGF, and HGF [31,32,33][31][32][33]. When STAT3 activation is induced in myeloid precursors, this factor controls cell survival, transcription of immunosuppressive enzymes (ARG1 and iNOS), prevents myeloid-cell maturation and results in aberrant differentiation into immature MDSCs [34]. Some of the key regulators of MDSC accumulation and activity are the transcription factors STAT3 and NF-κβ. STAT3 enhances MDSC accumulation through several pathways. STAT3 and STAT5 inhibit IRF8, a crucial transcription factor that drives normal myeloid differentiation into monocytes and dendritic cells, and down-regulates the differentiation of MDSCs, when this is necessary to inhibit their pathological expansion [35]. The proinflammatory damage-associated molecular pattern DAMP is commonly found in the TME and activates MDSC through NF-κβ. STAT3 upregulates p47phox and gp91, which increases •NO and peroxynitrite [1]. Peroxynitrite is formed when nitric oxide •NO reacts with superoxide anion •O2− [1] due to the overexpression of two subunits of NADPH oxidase, p47phox and gp91 (derived from phosphorylation of STAT3, a hallmark of MDSC) [32]. Peroxynitrite is an anion derived from the reaction of •NO with •O2−, Figure 1.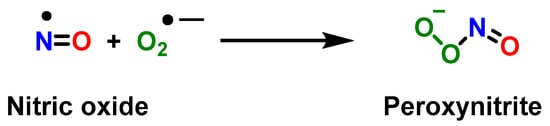
3. Role of Myeloid-Derived Suppressor Cells in Cancer
In the early 1970s, initial research was published linking tumor growth to the proliferation of immunosuppressive myeloid cells. Research conducted in the 1980s and 1990s by Diana Lopez, Jim Talmadge, M. Rita Young, and Hans Schreiber showed that different types of myeloid cells suppressed immunological function in tumor cell growth [3]. In most types of cancer, PMN-MDSCs account for more than 80% of MDSCs. There is another small group (less than 3%) of cells with myeloid colony-forming activity that represent a mixture of myeloid progenitors and precursors [40]. Myeloid-derived suppressor cells are present in virtually all cancer patients, impair adaptive and innate anti-tumor immunity, and promote tumor progression by non-immune mechanisms. Their widespread presence combined with their diverse peritumoral activities makes them a major obstacle to cancer immunotherapy [41]. MDSCs have been detected in cancer patients and mice with tumors for more than 30 years. They inhibit antitumor immunity and act through CBI-independent signaling pathways [42,43][42][43]. In addition, MDSCs interfere with antibody treatments and promote tumor development via a variety of non-immune pathways [44]. Tumor immunity represents a new avenue for improved cancer therapy. Evasion of the immune system is a key feature of tumors [45]. To successfully establish themselves in a host and continue to grow, tumor cells use biochemical signals to hide from the host’s immune response and remain undetected. Immunotherapy aims to restore the immune response and immunity to cancer and has revolutionized cancer therapy in recent years. However, immunosuppressive rogue cells such as tumor-associated macrophages TAM, tumor-associated neutrophils TAN, regulatory T cells Treg, regulatory dendritic cells RegDC, cancer-associated fibroblasts, and MDSCs remain a major obstacle to immunotherapy and contribute to treatment failure, reduced life expectancy, and poor prognosis [46,47,48][46][47][48]. Checkpoint blockade immunotherapy CBI has been a revolution in cancer treatment because the patient’s adaptive immune system can eradicate malignant cells once the immunosuppressive mechanisms are neutralized [43]. Immune checkpoint inhibitors have successfully improved outcomes in various tumor types, and immune cell-based therapy is also gaining attention [49]. However, this IBC treatment is only effective in a certain group of cancer patients, as other immunosuppressive mechanisms appear to block T-cell-induced anti-tumor immunity [50]. As seen, immunosuppression plays a crucial role in tumor progression and contributes to the frequent failure of immunotherapy treatments and potential cancer vaccines, so it is necessary to address the study of the inhibition of these MDSCs to ensure the viability of the cancer immunotherapy approach. Elimination of suppressor factors is now recognized as a necessary step toward effective cancer immunotherapy. Drugs such as gemcitabine can be used to completely remove MDSCs from the body. There was no appreciable decrease in the number of B and T cells, suggesting that this effect occurs only in MDSCs. In contrast, a study of 17 patients with early-stage breast cancer found that chemotherapy with doxorubicin and cyclophosphamide resulted in an increase in the number of MDSCs in the peripheral blood [51].4. Role of Myeloid-Derived Suppressor Cells in COVID-19
MDSCs have been described in a number of viral diseases, including respiratory infections [52]. It is still unclear how these cells contribute to the development of infectious diseases. On the one hand, MDSCs hinder the body’s ability to eliminate pathogens from the bloodstream and from the site of infection by suppressing the actions of effector-immune cells. On the other hand, MDSCs can prevent host organs from suffering lethal dysfunction by limiting the hyperinflammation and “cytokine storm” caused by infection [53]. During the infective process of COVD-19, pathogen-associated molecular patterns PAMPs from the replication of SARS-CoV-2 in host cells are recognized by a variety of membrane PRR pattern recognition receptors, including Toll-like receptors TLR-3, -4, -7, and -8; in addition to the porin domain of the NOD-like receptor family NLRP3, the retinoic acid-inducible RIG1, melanoma differentiation-associated protein 5 MDA5, and LGP2 are also present. Single-stranded RNA of SARS-CoV-2 is recognized by TLR-7 and -8, while double-stranded RNA intermediates are bound to TLR -3, RIG1, LGP2, and MDA5. SARS-CoV-2 proteins are recognized by TLR-4 and NLRP3 [54,55,56][54][55][56]. Moreover, viral replication triggers the synthesis of host-specific threat-associated molecular patterns DAMPs, which are then secreted extracellularly by injured or dying infected cells after rupture of the plasma membrane and recognized by cells bearing pattern recognition receptors PRRs. Calprotectin S100A8/A9, HMGB1 protein, mitochondrial DNA mt-DNA, and extracellular secreted nicotinamide phosphoribosyl transferase eNAMPT are important DAMPs associated with COVID-19 [57,58,59][57][58][59]. In response to PAMPs and DAMPs, many cell types secrete inflammatory mediators, chemokines, and growth factors such as interleukins IL-1B, IL-6, IFNα/β, TNF-α, chemokine ligand CXCL8/IL-8, CXCL10, chemokine ligand CCL5, granulocyte colony-stimulating factors G-CSF, and granulocyte-macrophage GM-CSF [60,61][60][61]. Recent studies point to a link between tissue damage and inflammation, in which the damage-associated molecular patterns of DAMPs play a key role in the etiology of severe COVID-19 [62]. NETs (neutrophil extracellular traps) are involved in the pathogenesis of COVID-19 and this can be seen in the fact that treatment of healthy neutrophils with serum from COVID-19 patients triggers NET release; and, in general, SARS-CoV-2 stimulates neutrophils to release NETs [63]. Several components of NETs, together with factors such as oxidative stress, contribute to the release of endogenous DAMPs, leading to severe hypoxia and ultimately acute respiratory distress syndrome ARDS in patients with severe COVID-19 [64]. The innate immune sets the adaptive response, which begins its activation within days [65]. Antigen-specific B cells, CD4+ T helper cells, and CD8+ cytotoxic T cells work together to orchestrate the adaptive response. However, CD4+ T helper cells are more prevalent than CD8+ cytotoxic T cells in SARS-CoV-2 [66] The sequencing of the immune response implies that innate immunity is activated first, followed by adaptive immunity, acting synchronously to eliminate the virus and damaged cells [67,68][67][68]. After the pathogen is eliminated, a series of immunoregulatory cell populations terminate the inflammatory response and restore tissue homeostasis. There is a multifactorial risk that can affect the course of COVID-19 disease, including hypertension, cancer, diabetes, as well as respiratory, cerebrovascular, and chronic kidney diseases [69]. These comorbidities are associated with an immunocompromised state characterized by an impaired immune response and decreased ability to fight viruses, as well as advanced age [70,71][70][71]. Myeloid cells play an important role in the pathogenesis of SARS-CoV-2, as evidenced by the frequent observation of a huge expansion of the myeloid-cell compartment and a decrease in the leukocyte compartment [72,73][72][73]. In COVID-19, myeloid cells are characterized by decreased antigen presentation and increased immunosuppressive characteristics, both of which are consistent with the profile of MDSC [73]. M2 macrophages, regulatory dendritic cells, regulatory T cells, and myeloid-derived suppressor cells are the primary immunoregulatory cell subsets that contribute to the attenuation of inflammation [53,74,75,76][53][74][75][76]. Although MDSCs can suppress a range of immune cells (including NK and B cells), their main goal is to induce T-cell immunosuppression. Therefore, evaluating their ability to block immune effector-cell activity is a critical component of understanding MDSCs [53,77,78][53][77][78]. In patients with COVID-19, MDSCs have been studied both in the peripheral blood and, more specifically, in the airways. As noted by Dean et al. 2021, large numbers of Arg1-expressing PMN-MDSCs were found in the lungs of COVID-19-deceased patients. This finding suggests that SARS-CoV-2 infection begins in the upper airways but progresses to the lower airways, where local recruitment of MDSCs is observed [79]. L-arginine is converted to ornithine via the enzyme arginase 1 Arg1, which inhibits T-cell proliferation and causes significant molecular changes in T cells, such as low CD3ζ chain expression and reduced IFN-γ production [80]. Interleukin IL -10 and transforming growth factor TGF-β produced by MDSCs inhibit T-cell activation and recruit regulatory Treg cells, respectively [81]. In addition, MDSCs can bind to PD1 molecules on T cells via their ligand PDL1 and induce T-cell death via cell-to-cell interactions [82]. PMN-MDSC generate ROS and nitric oxide •NO, and this allows the formation of peroxynitrite, which in turn is broken down to generate two new radicals, •NO2 and •OH, with a high oxidative capacity [83]. •NO2 induces T-cell receptor nitration on CD8+ cells (they are cytotoxic, like CD4+ T-helper cells, and express the TCR-cell receptor), during cell-to-cell contacts [84]. This nitration causes T cells to lose their ability to bind to the phosphorylated MHC (major histocompatibility complex) and therefore are unable to perform their function and respond to specific antigens, resulting in antigen-specific T cell tolerance [85]. Finally, according to a number of studies, there is a correlation between the number of MDSCs and the severity of COVID-19. Patients who required treatment in the ICU had more PMN-MDSCs than patients who did not, according to a 2020 study by Sacchi et al. [86]. In addition, Reizine et al. 2021 discovered that patients with acute respiratory distress syndrome ARDS have more PMN-MDSCs and M-MDSCs than individuals with moderate disease [87].References
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701.
- De Kleer, I.; Willems, F.; Lambrecht, B.; Goriely, S. Ontogeny of myeloid cells. Front. Immunol. 2014, 5, 423.
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752.
- Millrud, C.R.; Bergenfelz, C.; Leandersson, K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017, 8, 3649.
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835.
- Morales, J.K.; Kmieciak, M.; Knutson, K.L.; Bear, H.D.; Manjili, M.H. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1-bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res. Treat. 2010, 123, 39–49.
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T. INAUGURAL ARTICLE by a Recently Elected Academy Member: Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+ Ly6C+ granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248.
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174.
- Serafini, P.; Bronte, V. Myeloid-derived suppressor cells in tumor-induced T cell suppression and tolerance. In Tumor-Induced Immune Suppression; Springer: Berlin/Heidelberg, Germany, 2014; pp. 99–150.
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+ CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421.
- Cassetta, L.; Bruderek, K.; Skrzeczynska-Moncznik, J.; Osiecka, O.; Hu, X.; Rundgren, I.M.; Lin, A.; Santegoets, K.; Horzum, U.; Godinho-Santos, A. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J. Immunother. Cancer 2020, 8, e001223.
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-derived suppressor cells: Immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol. 2018, 200, 422–431.
- Gimeno, R.; Barquinero, J. Myeloid-derived suppressor cells (MDSC): Another player in the orchestra. Inmunología 2011, 30, 45–53.
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110.
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498.
- Lang, S.; Bruderek, K.; Kaspar, C.; Höing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell SubsetsCD16+/CD11b+ PMN-MDSC in Head and Neck Cancer. Clin. Cancer Res. 2018, 24, 4834–4844.
- Sarkar, T.; Dhar, S.; Sa, G. Tumor-infiltrating T-regulatory cells adapt to altered metabolism to promote tumor-immune escape. Curr. Res. Immunol. 2021, 2, 132–141.
- Burke, M.; Choksawangkarn, W.; Edwards, N.; Ostrand-Rosenberg, S.; Fenselau, C. Exosomes from myeloid-derived suppressor cells carry biologically active proteins. J. Proteome Res. 2014, 13, 836–843.
- Bronte, V.; Chappell, D.B.; Apolloni, E.; Cabrelle, A.; Wang, M.; Hwu, P.; Restifo, N.P. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J. Immunol. 1999, 162, 5728–5737.
- Waight, J.D.; Hu, Q.; Miller, A.; Liu, S.; Abrams, S.I. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS ONE 2011, 6, e27690.
- Raber, P.; Ochoa, A.C.; Rodríguez, P.C. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: Mechanisms of T cell suppression and therapeutic perspectives. Immunol. Investig. 2012, 41, 614–634.
- Koehn, B.H.; Apostolova, P.; Haverkamp, J.M.; Miller, J.S.; McCullar, V.; Tolar, J.; Munn, D.H.; Murphy, W.J.; Brickey, W.J.; Serody, J.S.; et al. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood 2015, 126, 1621–1628.
- Marigo, I.; Bosio, E.; Solito, S.; Mesa, C.; Fernandez, A.; Dolcetti, L.; Ugel, S.; Sonda, N.; Bicciato, S.; Falisi, E.; et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010, 32, 790–802.
- Ku, A.W.; Muhitch, J.B.; Powers, C.A.; Diehl, M.; Kim, M.; Fisher, D.T.; Sharda, A.P.; Clements, V.K.; O’Loughlin, K.; Minderman, H. Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymph nodes. Elife 2016, 5, e17375.
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807.
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of Tumor-Derived Prostaglandin-E2 Blocks the Induction of Myeloid-Derived Suppressor Cells and Recovers Natural Killer Cell ActivityRescue of NK Cells by Blocking the Induction of MDSCs. Clin. Cancer Res. 2014, 20, 4096–4106.
- Tsukamoto, H.; Nishikata, R.; Senju, S.; Nishimura, Y. Myeloid-Derived Suppressor Cells Attenuate TH1 Development through IL-6 Production to Promote Tumor ProgressionDampening of Antitumor TH1 Development by MDSC-Derived IL-6. Cancer Immunol. Res. 2013, 1, 64–76.
- Pan, P.-Y.; Ma, G.; Weber, K.J.; Ozao-Choy, J.; Wang, G.; Yin, B.; Divino, C.M.; Chen, S.-H. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010, 70, 99–108.
- Kwak, T.; Wang, F.; Deng, H.; Condamine, T.; Kumar, V.; Perego, M.; Kossenkov, A.; Montaner, L.J.; Xu, X.; Xu, W. Distinct populations of immune-suppressive macrophages differentiate from monocytic myeloid-derived suppressor cells in cancer. Cell Rep. 2020, 33, 108571.
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453.
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51.
- Su, Y.-L.; Banerjee, S.; White, S.V.; Kortylewski, M. STAT3 in tumor-associated myeloid cells: Multitasking to disrupt immunity. Int. J. Mol. Sci. 2018, 19, 1803.
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185.
- Fultang, N.; Li, X.; Li, T.; Chen, Y.H. Myeloid-derived suppressor cell differentiation in cancer: Transcriptional regulators and enhanceosome-mediated mechanisms. Front. Immunol. 2021, 11, 619253.
- Waight, J.D.; Netherby, C.; Hensen, M.L.; Miller, A.; Hu, Q.; Liu, S.; Bogner, P.N.; Farren, M.R.; Lee, K.P.; Liu, K. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J. Clin. Investig. 2013, 123, 4464–4478.
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962.
- Lapurga, G.; Sun, S.; Carlson, E.; Savardekar, H.; Kendra, K.; Peterson, B.; Carson, W. 686 Characterization of a novel compound that inhibits peroxynitrite generation by myeloid derived suppressor cells. J. Immunother. Cancer 2021, 9, A714.
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802.
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244.
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150.
- Park, S.M.; Youn, J.I. Role of myeloid-derived suppressor cells in immune checkpoint inhibitor therapy in cancer. Arch. Pharmacal Res. 2019, 42, 560–566.
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra67.
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; Andreu-Agullo, C.; Derbyshire, M.L.; Posada, J.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840.e18.
- Horikawa, N.; Abiko, K.; Matsumura, N.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Murakami, R.; Taki, M.; Ukita, M.; Hosoe, Y.; et al. Anti-VEGF therapy resistance in ovarian cancer is caused by GM-CSF-induced myeloid-derived suppressor cell recruitment. Br. J. Cancer 2020, 122, 778–788.
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198.
- Mantovani, A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur. J. Immunol. 2010, 40, 3317–3320.
- Yang, Z.Z.; Ansell, S.M. The role of Treg cells in the cancer immunological response. Am. J. Immunol. 2009, 5, 17–28.
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36.
- Tsuchiya, H.; Shiota, G. Immune evasion by cancer stem cells. Regen. Ther. 2021, 17, 20–33.
- Gabrilovich, D.I.; Bronte, V.; Chen, S.-H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425.
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59.
- Koushki, K.; Salemi, M.; Miri, S.M.; Arjeini, Y.; Keshavarz, M.; Ghaemi, A. Role of myeloid-derived suppressor cells in viral respiratory infections; hints for discovering therapeutic targets for COVID-19. Biomed. Pharmacother. 2021, 144, 112346.
- Fullerton, J.N.; O’Brien, A.J.; Gilroy, D.W. Pathways mediating resolution of inflammation: When enough is too much. J. Pathol. 2013, 231, 8–20.
- Chen, I.-Y.; Moriyama, M.; Chang, M.-F.; Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019, 10, 50.
- Yamada, T.; Sato, S.; Sotoyama, Y.; Orba, Y.; Sawa, H.; Yamauchi, H.; Sasaki, M.; Takaoka, A. RIG-I triggers a signaling-abortive anti-SARS-CoV-2 defense in human lung cells. Nat. Immunol. 2021, 22, 820–828.
- Yin, X.; Riva, L.; Pu, Y.; Martin-Sancho, L.; Kanamune, J.; Yamamoto, Y.; Sakai, K.; Gotoh, S.; Miorin, L.; De Jesus, P.D. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep. 2021, 34, 108628.
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 992–994.
- Scozzi, D.; Cano, M.; Ma, L.; Zhou, D.; Zhu, J.H.; O’Halloran, J.A.; Goss, C.; Rauseo, A.M.; Liu, Z.; Sahu, S.K. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 2021, 6, e143299.
- Bime, C.; Casanova, N.G.; Nikolich-Zugich, J.; Knox, K.S.; Camp, S.M.; Garcia, J.G. Strategies to DAMPen COVID-19-mediated lung and systemic inflammation and vascular injury. Transl. Res. 2021, 232, 37–48.
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353.
- Land, W.G. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome—With a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. 2021, 22, 141–160.
- Parthasarathy, U.; Martinelli, R.; Vollmann, E.H.; Best, K.; Therien, A.G. The impact of DAMP-mediated inflammation in severe COVID-19 and related disorders. Biochem. Pharmacol. 2022, 195, 114847.
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999.
- Grasselli, G.; Tonetti, T.; Protti, A.; Langer, T.; Girardis, M.; Bellani, G.; Laffey, J.; Carrafiello, G.; Carsana, L.; Rizzuto, C. Pathophysiology of COVID-19-associated acute respiratory distress syndrome: A multicentre prospective observational study. Lancet Respir. Med. 2020, 8, 1201–1208.
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859.
- Moderbacher, C.R.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell 2020, 183, 996–1012.e19.
- Shah, V.K.; Firmal, P.; Alam, A.; Ganguly, D.; Chattopadhyay, S. Overview of immune response during SARS-CoV-2 infection: Lessons from the past. Front. Immunol. 2020, 11, 1949.
- Anand, U.; Jakhmola, S.; Indari, O.; Jha, H.C.; Chen, Z.-S.; Tripathi, V.; de la Lastra, J.M.P. Potential therapeutic targets and vaccine development for SARS-CoV-2/COVID-19 pandemic management: A review on the recent update. Front. Immunol. 2021, 12, 658519.
- Gold, M.S.; Sehayek, D.; Gabrielli, S.; Zhang, X.; McCusker, C.; Ben-Shoshan, M. COVID-19 and comorbidities: A systematic review and meta-analysis. Postgrad. Med. 2020, 132, 749–755.
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, immunity, and COVID-19: How age influences the host immune response to coronavirus infections? Front. Physiol. 2021, 11, 571416.
- Latif, M.B.; Shukla, S.; Estrada, P.M.d.R.; Ribeiro, S.P.; Sekaly, R.P.; Sharma, A.A. Immune mechanisms in cancer patients that lead to poor outcomes of SARS-CoV-2 infection. Transl. Res. 2022, 241, 83–95.
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362.
- Tang, G.; Huang, M.; Luo, Y.; Liu, W.; Lin, Q.; Mao, L.; Wu, S.; Xiong, Z.; Hou, H.; Sun, Z. The dynamic immunological parameter landscape in coronavirus disease 2019 patients with different outcomes. Front. Immunol. 2021, 12, 697622.
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 2020, 181, 1489–1501.
- Zhao, J.; Yuan, Q.; Wang, H.; Liu, W.; Liao, X.; Su, Y.; Wang, X.; Yuan, J.; Li, T.; Li, J. Antibody responses to SARS-CoV-2 in patients with novel coronavirus disease 2019. Clin. Infect. Dis. 2020, 71, 2027–2034.
- Suthar, M.S.; Zimmerman, M.G.; Kauffman, R.C.; Mantus, G.; Linderman, S.L.; Hudson, W.H.; Vanderheiden, A.; Nyhoff, L.; Davis, C.W.; Adekunle, O. Rapid generation of neutralizing antibody responses in COVID-19 patients. Cell Rep. Med. 2020, 1, 100040.
- Özkan, B.; Lim, H.; Park, S.-G. Immunomodulatory function of myeloid-derived suppressor cells during B cell-mediated immune responses. Int. J. Mol. Sci. 2018, 19, 1468.
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: Roles in tumor progression. Front. Immunol. 2019, 10, 771.
- Dean, M.J.; Ochoa, J.B.; Sanchez-Pino, M.; Zabaleta, J.; Garai, J.; Del Valle, L.; Wyczechowska, D.; Buckner, L.; Philbrook, P.; Majumder, R. Transcriptome and functions of granulocytic myeloid-derived suppressor cells determine their association with disease severity of COVID-19. medRxiv 2021.
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573.
- Pyzer, A.R.; Cole, L.; Rosenblatt, J.; Avigan, D.E. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int. J. Cancer 2016, 139, 1915–1926.
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790.
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642.
- Ohl, K.; Tenbrock, K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front. Immunol. 2018, 9, 2499.
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835.
- Sacchi, A.; Grassi, G.; Bordoni, V.; Lorenzini, P.; Cimini, E.; Casetti, R.; Tartaglia, E.; Marchioni, L.; Petrosillo, N.; Palmieri, F. Early expansion of myeloid-derived suppressor cells inhibits SARS-CoV-2 specific T-cell response and may predict fatal COVID-19 outcome. Cell Death Dis. 2020, 11, 921.
- Reizine, F.; Lesouhaitier, M.; Gregoire, M.; Pinceaux, K.; Gacouin, A.; Maamar, A.; Painvin, B.; Camus, C.; Le Tulzo, Y.; Tattevin, P. SARS-CoV-2-induced ARDS associates with MDSC expansion, lymphocyte dysfunction, and arginine shortage. J. Clin. Immunol. 2021, 41, 515–525.