 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hanna Heloterä | -- | 3760 | 2023-02-01 08:31:17 | | | |
| 2 | Lindsay Dong | Meta information modification | 3760 | 2023-02-02 01:44:54 | | |
Video Upload Options
Age-related macular degeneration (AMD) is the leading cause of visual impairment in the aging population with a limited understanding of its pathogenesis and the number of patients are all the time increasing. AMD is classified into two main forms: dry and neovascular AMD (nAMD). Dry AMD is the most prevalent form (80–90%) of AMD cases. Neovascular AMD (10–20% of AMD cases) is treated with monthly or more sparsely given intravitreal anti-vascular endothelial growth factor inhibitors, but unfortunately, not all patients respond to the current treatments. A clinical hallmark of nAMD is choroidal neovascularization. The progression of AMD is initially characterized by atrophic alterations in the retinal pigment epithelium, as well as the formation of lysosomal lipofuscin and extracellular drusen deposits. Cellular damage caused by chronic oxidative stress, protein aggregation and inflammatory processes may lead to advanced geographic atrophy and/or choroidal neovascularization and fibrosis.
1. Introduction
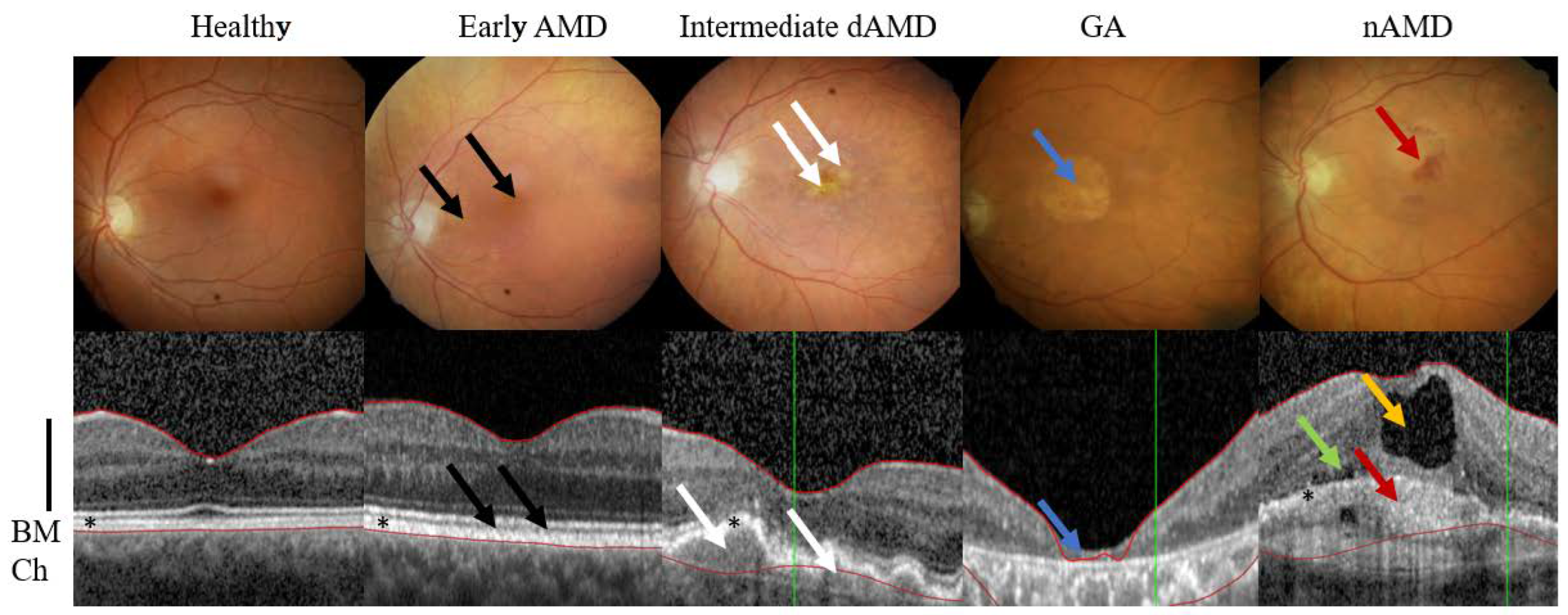
2. AMD Phenotypes
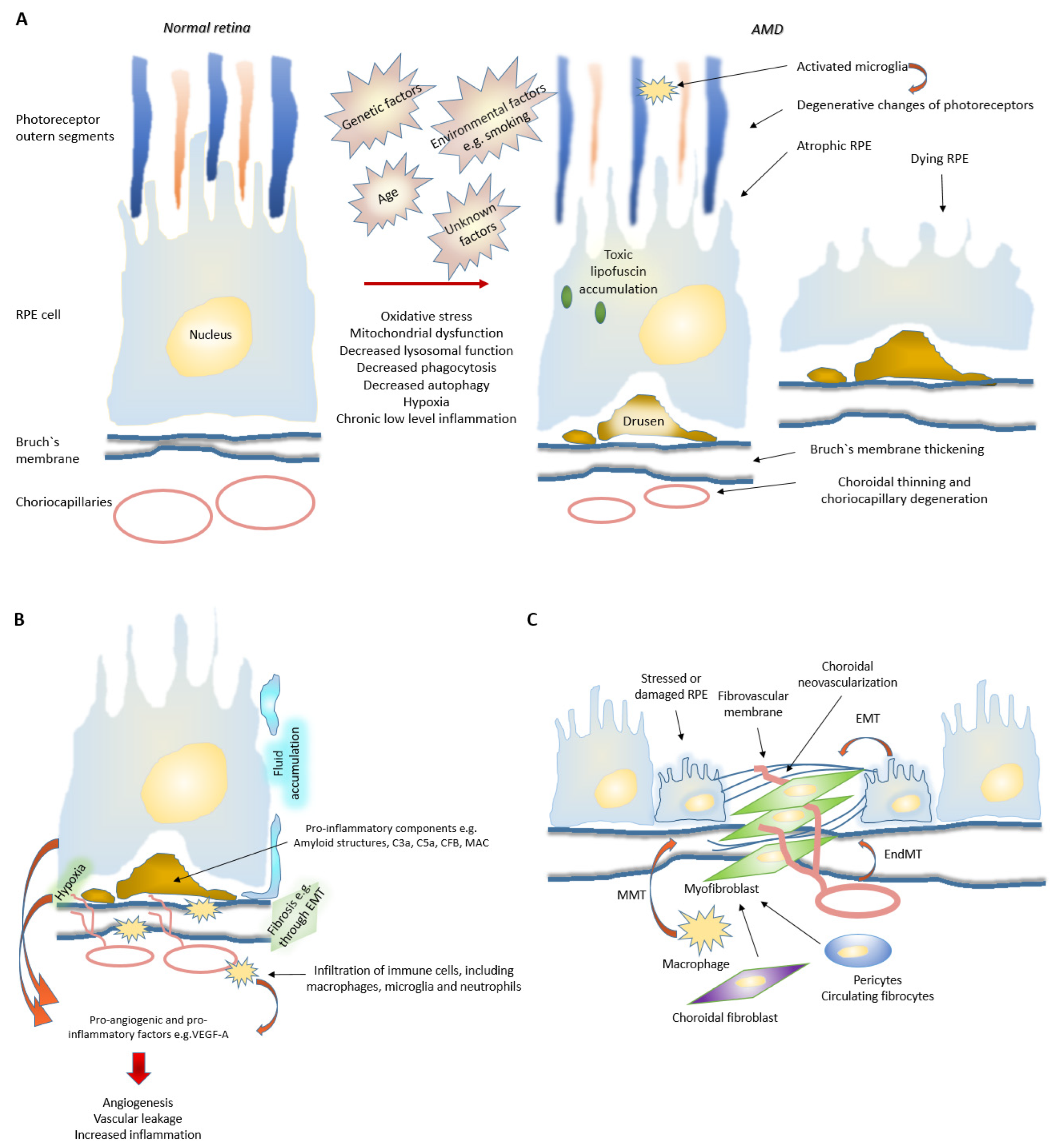
3. Evolution of AMD
3.1. Disturbed Proteostasis in AMD
The exact mechanism behind RPE degeneration and the onset and progression of AMD are not fully understood. However, dysfunction of many cellular processes, such as mitochondrial dysfunction, increased reactive oxygen species (ROS) production and protein aggregation, impaired autophagy and chronic inflammation have been linked to the development of AMD (Figure 2) [6][7][8][9][10][11].In general, autophagy is a self-clearance pathway that removes dysfunctional cellular components through lysosomal dependent mechanism and thus protects retinal cells from e.g., oxidative stress induced insults [7][8][10]. Interestingly, hypoxia, which is also a strong driver of angiogenesis, has been shown to induce autophagy, which in turn might act as a survival mechanism for hypoxic cells through recycling of cellular constituents [12][13]. Dysfunctional lysosomal clearance and accumulation of waste materials play key roles in cellular levels during AMD development. Autophagy is especially important for normal RPE homeostasis as RPE cells have undergone terminal differentiation and do not divide anymore or divide only rarely. One of the key roles of RPE cells is phagocytosis of lipid-rich photoreceptor outer segments (POS). POS are degraded in lysosomes. This process is called heterophagy that should be differentiated from autophagy, although lysosomes are regulating both processes. Accumulated lipofuscin during AMD development disturbs both processes heterophagy and autophagy [14]. Oxidative stress is known to induce autophagic flux in RPE cells, but chronic oxidative stress seems to lead to increased lysosomal lipofuscin but decreased lysosomal activity and autophagy. While these processes play an important role in the homeostasis of RPE cells, their impairment can lead to detrimental accumulation of damaged organelles and abnormal or toxic proteins [7][10]. Disturbed proteostasis is clearly demonstrated by the accumulation of lysosomal lipofuscin in the RPE and extracellular drusen deposits between the RPE and choriocapillaris [6]. It is shown that lysosomal degradative functions can be further inhibited in RPE cells by a lipofuscin, and accumulation of lipofuscin in the RPE is a sign of cellular senescence in AMD. Intralysosomal accumulation of toxic lipofuscin can also sensitize lysosomes to visible light, oxidative stress, jeopardize lysosomal stability and lead to cellular apoptosis [15].
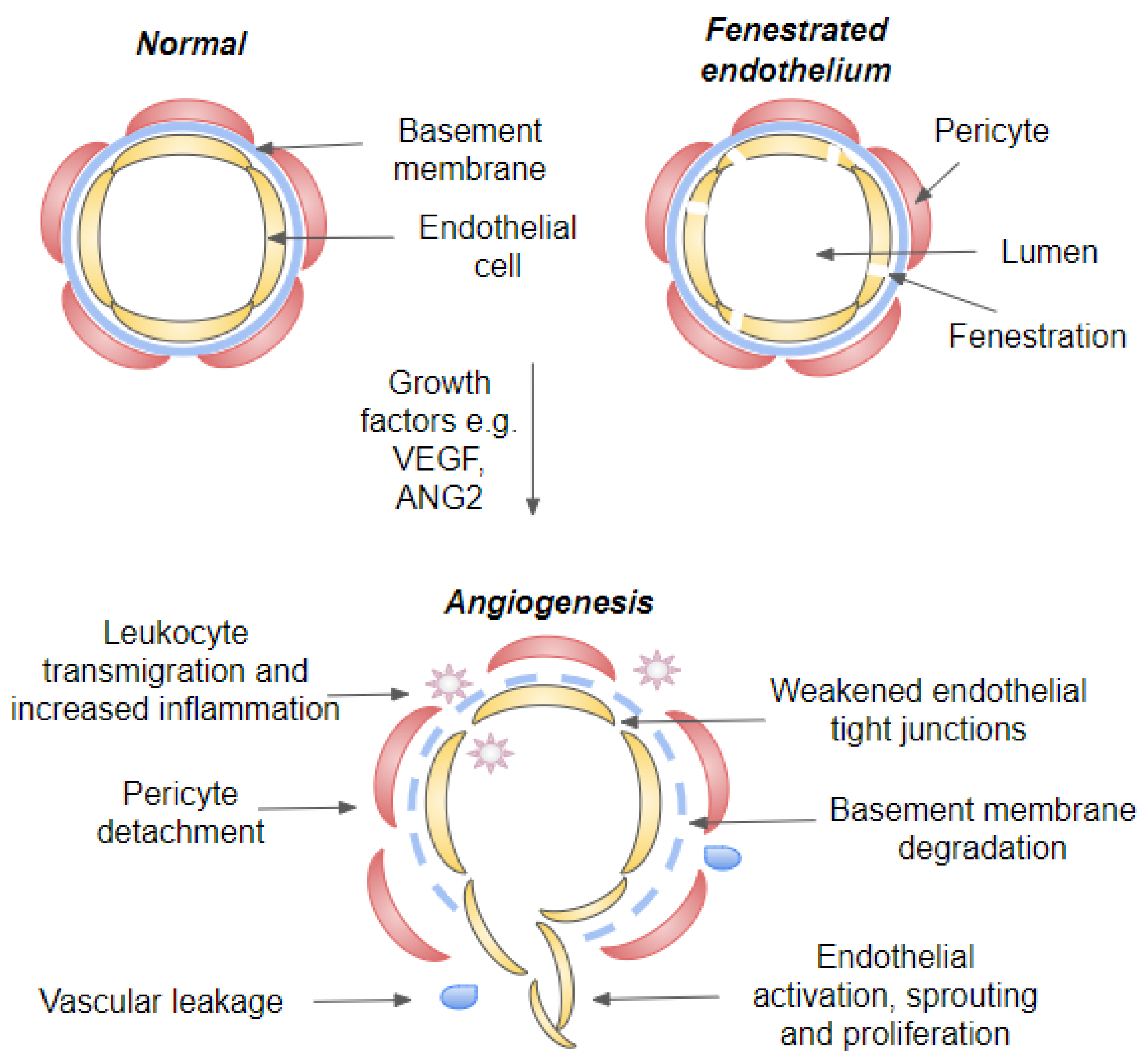
3.2. Role of Choriocapillaris in nAMD Development
3.3. Senescence in AMD
During AMD progression, decreased autophagy is also associated with cellular senescence and senescence-associated secretory phenotype is associated with the release of ROS, selective growth factors, and inflammatory cytokines, chemokines and proteases. Senescent cells also promote the senescence of surrounding cells’ secretory phenotype, which can contribute to the maintenance of a chronic state of low-grade inflammation in tissues and organs. They are also apoptosis resistant, failing to enter programmed cell death and rather aggregate instead. Therefore, they may regulate drusen biogenesis and advanced GA and nAMD development [19][20]. Term immunosenescence is used to describe altered immune functions during aging.
3.4. Fibrosis in nAMD
4. Angiogenesis and Inflammation
4.1. Inflammatory Signaling Cascades
4.2. Blood-Retinal-Barriers
4.3. Inflammatory Cell Recruitment
Already 1985 Penfold et al., described the involvement of immunocompetent cells in early, intermediate and late stage of AMD. They suggested that macrophages, fibroblast, lymphocytes and mast cells play a role in neovascularization, atrophy of RPE and Bruch`s membrane breakdown [41]. During the inflammation process, leukocytes sense the chemokine gradient originating from the inflamed tissue and begin to make contact with the adhesion molecules expressed by endothelial cells to permit their tighter binding to the vascular endothelium. Finally, leukocytes leave the circulation by following the chemokine gradient and move towards the damaged tissue, where they become activated [28]. Studies done with an inducible model of photoreceptor death in mice showed infiltration of 12 distinct subpopulations of microglia, monocytes and macrophages [42]. Previously mentioned study did not identify markers for neutrophils, but infiltration of neutrophils has been described from early AMD patient samples and studies done with mouse models suggested the role of neutrophils in retinal degenerations [42][43].4.4. Effects of Hypoxia
References
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990-2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234.
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116.
- Li, J.Q.; Welchowski, T.; Schmid, M.; Mauschitz, M.M.; Holz, F.G.; Finger, R.P. Prevalence and incidence of age-related macular degeneration in europe: A systematic review and meta-analysis. Br. J. Ophthalmol. 2020, 104, 1077–1084.
- Flaxel, C.J.; Adelman, R.A.; Bailey, S.T.; Fawzi, A.; Lim, J.I.; Vemulakonda, G.A.; Ying, G.S. Age-related macular degeneration preferred practice pattern(r). Ophthalmology 2020, 127, P1–P65.
- Moon, B.G.; Joe, S.G.; Hwang, J.U.; Kim, H.K.; Choe, J.; Yoon, Y.H. Prevalence and risk factors of early-stage age-related macular degeneration in patients examined at a health promotion center in korea. J. Korean Med. Sci. 2012, 27, 537–541.
- Anderson, D.H.; Mullins, R.F.; Hageman, G.S.; Johnson, L.V. A role for local inflammation in the formation of drusen in the aging eye. Am. J. Ophthalmol. 2002, 134, 411–431.
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell Mol. Life Sci. 2016, 73, 1765–1786.
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the rpe is associated with increased susceptibility to oxidative stress and amd. Autophagy 2014, 10, 1989–2005.
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med. 2008, 14, 194–198.
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858.
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311.
- Du, H.; Yang, W.; Chen, L.; Shen, B.; Peng, C.; Li, H.; Ann, D.K.; Yen, Y.; Qiu, W. Emerging role of autophagy during ischemia-hypoxia and reperfusion in hepatocellular carcinoma. Int. J. Oncol. 2012, 40, 2049–2057.
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133.
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984.
- Moreno-Garcia, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An overview of the role of lipofuscin in age-related neurodegeneration. Front Neurosci. 2018, 12, 464.
- Stefansson, E.; Geirsdottir, A.; Sigurdsson, H. Metabolic physiology in age related macular degeneration. Prog. Retin. Eye Res. 2011, 30, 72–80.
- Lipecz, A.; Csipo, T.; Tarantini, S.; Hand, R.A.; Ngo, B.N.; Conley, S.; Nemeth, G.; Tsorbatzoglou, A.; Courtney, D.L.; Yabluchanska, V.; et al. Age-related impairment of neurovascular coupling responses: A dynamic vessel analysis (dva)-based approach to measure decreased flicker light stimulus-induced retinal arteriolar dilation in healthy older adults. Geroscience 2019, 41, 341–349.
- McHugh, K.J.; Li, D.; Wang, J.C.; Kwark, L.; Loo, J.; Macha, V.; Farsiu, S.; Kim, L.A.; Saint-Geniez, M. Computational modeling of retinal hypoxia and photoreceptor degeneration in patients with age-related macular degeneration. PLoS ONE 2019, 14, e0216215.
- Wang, S.; Wang, X.; Cheng, Y.; Ouyang, W.; Sang, X.; Liu, J.; Su, Y.; Liu, Y.; Li, C.; Yang, L.; et al. Autophagy dysfunction, cellular senescence, and abnormal immune-inflammatory responses in amd: From mechanisms to therapeutic potential. Oxid. Med. Cell Longev. 2019, 2019, 3632169.
- Kaarniranta, K.; Blasiak, J.; Liton, P.; Boulton, M.; Klionsky, D.J.; Sinha, D. Autophagy in age-related macular degeneration. Autophagy 2022, 1–13.
- Little, K.; Ma, J.H.; Yang, N.; Chen, M.; Xu, H. Myofibroblasts in macular fibrosis secondary to neovascular age-related macular degeneration-the potential sources and molecular cues for their recruitment and activation. EBioMedicine 2018, 38, 283–291.
- Tenbrock, L.; Wolf, J.; Boneva, S.; Schlecht, A.; Agostini, H.; Wieghofer, P.; Schlunck, G.; Lange, C. Subretinal fibrosis in neovascular age-related macular degeneration: Current concepts, therapeutic avenues, and future perspectives. Cell Tissue Res. 2022, 387, 361–375.
- Friedlander, M. Fibrosis and diseases of the eye. J. Clin. Investig. 2007, 117, 576–586.
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K. Emt: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94.
- Li, H. Angiogenesis in the progression from liver fibrosis to cirrhosis and hepatocelluar carcinoma. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 217–233.
- Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis updated. Circ. Res. 2020, 127, 310–329.
- Jeong, J.H.; Ojha, U.; Lee, Y.M. Pathological angiogenesis and inflammation in tissues. Arch. Pharm. Res. 2021, 44, 1–15.
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435.
- Aguilar-Cazares, D.; Chavez-Dominguez, R.; Carlos-Reyes, A.; Lopez-Camarillo, C.; Hernadez de la Cruz, O.N.; Lopez-Gonzalez, J.S. Contribution of angiogenesis to inflammation and cancer. Front. Oncol. 2019, 9, 1399.
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815.
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the endothelial barrier: Identifying and reconciling controversies. Trends Mol. Med. 2021, 27, 314–331.
- O’Leary, F.; Campbell, M. The blood-retina barrier in health and disease. FEBS J. 2021. Published Online Ahead of Print.
- Chanchal, S.; Mishra, A.; Singh, M.K.; Ashraf, M.Z. Understanding inflammatory responses in the manifestation of prothrombotic phenotypes. Front Cell Dev. Biol. 2020, 8, 73.
- Tisi, A.; Feligioni, M.; Passacantando, M.; Ciancaglini, M.; Maccarone, R. The impact of oxidative stress on blood-retinal barrier physiology in age-related macular degeneration. Cells 2021, 10, 64.
- Marneros, A.G.; Fan, J.; Yokoyama, Y.; Gerber, H.P.; Ferrara, N.; Crouch, R.K.; Olsen, B.R. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am. J. Pathol. 2005, 167, 1451–1459.
- Kim, S.A.; Kim, S.J.; Choi, Y.A.; Yoon, H.J.; Kim, A.; Lee, J. Retinal vegfa maintains the ultrastructure and function of choriocapillaris by preserving the endothelial plvap. Biochem. Biophys. Res. Commun. 2020, 522, 240–246.
- Kamba, T.; Tam, B.Y.; Hashizume, H.; Haskell, A.; Sennino, B.; Mancuso, M.R.; Norberg, S.M.; O’Brien, S.M.; Davis, R.B.; Gowen, L.C.; et al. Vegf-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H560–H576.
- Izzedine, H.; Escudier, B.; Lhomme, C.; Pautier, P.; Rouvier, P.; Gueutin, V.; Baumelou, A.; Derosa, L.; Bahleda, R.; Hollebecque, A.; et al. Kidney diseases associated with anti-vascular endothelial growth factor (vegf): An 8-year observational study at a single center. Medicine 2014, 93, 333–339.
- Ollero, M.; Sahali, D. Inhibition of the vegf signalling pathway and glomerular disorders. Nephrol. Dial. Transpl. 2015, 30, 1449–1455.
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of vegf-a expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716.
- Penfold, P.L.; Killingsworth, M.C.; Sarks, S.H. Senile macular degeneration: The involvement of immunocompetent cells. Graefes Arch. Clin. Exp. Ophthalmol. 1985, 223, 69–76.
- Ronning, K.E.; Karlen, S.J.; Miller, E.B.; Burns, M.E. Molecular profiling of resident and infiltrating mononuclear phagocytes during rapid adult retinal degeneration using single-cell rna sequencing. Sci. Rep. 2019, 9, 4858.
- Ghosh, S.; Padmanabhan, A.; Vaidya, T.; Watson, A.M.; Bhutto, I.A.; Hose, S.; Shang, P.; Stepicheva, N.; Yazdankhah, M.; Weiss, J.; et al. Neutrophils homing into the retina trigger pathology in early age-related macular degeneration. Commun. Biol. 2019, 2, 348.
- Buschini, E.; Piras, A.; Nuzzi, R.; Vercelli, A. Age related macular degeneration and drusen: Neuroinflammation in the retina. Prog. Neurobiol. 2011, 95, 14–25.
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611.
- Droho, S.; Cuda, C.M.; Perlman, H.; Lavine, J.A. Macrophage-derived interleukin-6 is necessary and sufficient for choroidal angiogenesis. Sci. Rep. 2021, 11, 18084.
- Baluk, P.; Yao, L.C.; Feng, J.; Romano, T.; Jung, S.S.; Schreiter, J.L.; Yan, L.; Shealy, D.J.; McDonald, D.M. Tnf-alpha drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J. Clin. Investig. 2009, 119, 2954–2964.
- Fahey, E.; Doyle, S.L. Il-1 family cytokine regulation of vascular permeability and angiogenesis. Front. Immunol. 2019, 10, 1426.
- Heidemann, J.; Ogawa, H.; Dwinell, M.B.; Rafiee, P.; Maaser, C.; Gockel, H.R.; Otterson, M.F.; Ota, D.M.; Lugering, N.; Domschke, W.; et al. Angiogenic effects of interleukin 8 (cxcl8) in human intestinal microvascular endothelial cells are mediated by cxcr2. J. Biol. Chem. 2003, 278, 8508–8515.
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257.



