2. AMD Phenotypes
AMD is a complex and multifactorial disease with linked etiologies such as age, genetic factors, cardiovascular diseases, smoking and unhealthy diet
[5][6][7][8][34,35,36,37]. It is a progressive disease, where degeneration of the retinal pigment epithelium (RPE) results in the death of photoreceptors, which in turn leads to a loss of central vision. Aging RPE faces oxidative stress, a factor that together with deteriorating functionality, e.g., decreased intracellular recycling and degradation of proteins that induces inflammation (
Figure 23)
[7][8][9][36,37,38]. Severity of the symptoms guides the disease classification and AMD can be divided into early, intermediate, and advanced forms (
Figure 1)
[4].
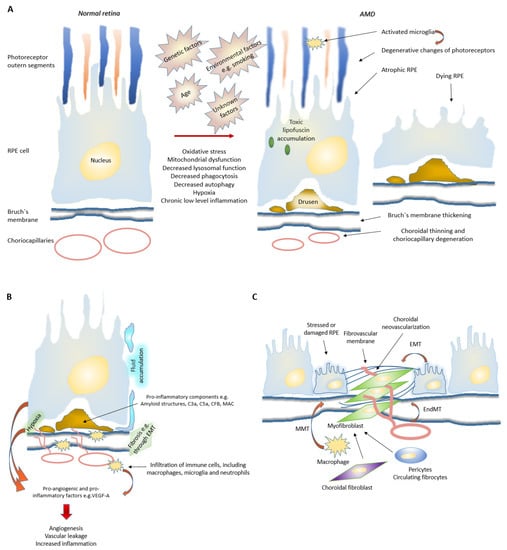
Figure 23. Mechanisms behind age related macular degeneration. (A) Age, light exposure, smoking, high-fat diet and unknown factors contribute to elevated oxidative stress, mitochondrial dysfunctions as well as decreased phagocytosis, autophagy and toxin clearance mechanisms in RPE cells. This leads to accumulation of intra- and extra-cellular waste products such as intracellular light absorbing lipofuscin and extracellular drusens. Affected retina may eventually face structural changes such as, RPE degeneration, photoreceptor loss, BrM thickening, choriocapillary degeneration, low-level inflammation and fibrosis. Activated microglia migrate to the outer nuclear layer to remove rod cell debris and, at the same time, may kill adjacent photoreceptors. (B) During an angiogenic switch, inflammatory cell recruitment is initiated, and as newly formed vessels are leaky, they contribute to retinal edema, hemorrhage and therefore further potentiate the pathological wound healing response. In nAMD, there exist many mechanisms how neovascularization and low-level inflammation can be stimulated. For example, damaged RPE produces excessive amounts of growth factors, which stimulate vascular growth. In addition, BrM thickening and drusens increase distance of RPE cells from choroidal vasculature and thus increases hypoxia, which in turn will upregulate pro-angiogenic and pro-inflammatory growth factors. Interestingly, drusens also consist of pro-inflammatory components, such as amyloid structures and C3a. Furthermore, accumulating immune cells may secrete inflammatory cytokines and angiogenic growth factors and therefore support the disease progression. (C) In nAMD, fibrosis is the product of defective and excessive wound healing response. It is believed that pro-inflammatory cytokines can promote differentiation and activation of matrix-producing myofibroblasts (e.g., EndMT and EMT). Multiple cell types, including fibroblast, fibrocytes, macrophages, RPE cells and endothelial cells, may potentially participate in this process. Mesenchymal cells in subretinal fibrotic lesions can for example, originate from the retinal pigment epithelium and/or choroidal endothelial cells through EMT and EndMT. In addition, macrophages are able to transdifferentiate to myofibroblasts through MMT. RPE cells and infiltrating macrophages are believed to be a major source of these cytokines. Moreover, hypoxia may promote EMT in RPE cells. Abbreviations: RPE, retinal pigment epithelium; EMT, epithelial to mesenchymal transition; C3a, complement component 3a; C5a, complement component 5a; CFB, complement factor B; MAC, membrane attack complex; VEGF-A, vascular endothelial growth factor A; EMT, epithelial–mesenchymal transition; EndMT, endothelial–mesenchymal transition; MMT, macrophage-mesenchyman-transition.
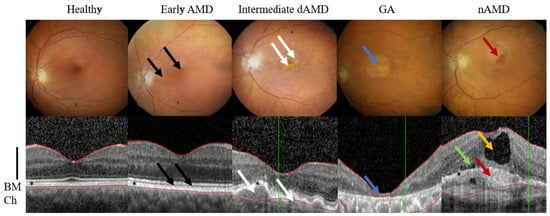
Early AMD is usually asymptomatic, even though accumulation of intracellular lipofuscin in the RPE and small extracellular drusens between RPE cells and Bruch’s membrane can be clinically detected (
Figure 1). Instead, intermediate AMD is characterized by an accumulation of multiple medium-sized or at least one big drusen (>125 μm). The advanced AMD is subdivided into two types of GA and nAMD. Term GA is used to describe areas where RPE cells and secondarily photoreceptors have completely damaged or died. Early, intermediate and GA forms consider only dry AMD, while choroidal neovascularization is a clinical hallmark in nAMD. Individuals with advanced AMD often have serious changes in their near and far vision. A functional degeneration of the RPE results in damage of photoreceptors and impaired maintenance of the sensory retina that led to loss of accurate vision and color detection. Thus, AMD greatly impairs the ability of an elderly patient to lead an independent life
[4].
3. Evolution of AMD
3.1. Disturbed Proteostasis in AMD
The exact mechanism behind RPE degeneration and the onset and progression of AMD are not fully understood. However, dysfunction of many cellular processes, such as mitochondrial dysfunction, increased reactive oxygen species (ROS) production and protein aggregation, impaired autophagy and chronic inflammation have been linked to the development of AMD (Figure 23) [6][7][8][9][10][11][35,36,37,38,39,40].In general, autophagy is a self-clearance pathway that removes dysfunctional cellular components through lysosomal dependent mechanism and thus protects retinal cells from e.g., oxidative stress induced insults [7][8][10][36,37,39]. Interestingly, hypoxia, which is also a strong driver of angiogenesis, has been shown to induce autophagy, which in turn might act as a survival mechanism for hypoxic cells through recycling of cellular constituents [12][13][41,42]. Dysfunctional lysosomal clearance and accumulation of waste materials play key roles in cellular levels during AMD development. Autophagy is especially important for normal RPE homeostasis as RPE cells have undergone terminal differentiation and do not divide anymore or divide only rarely. One of the key roles of RPE cells is phagocytosis of lipid-rich photoreceptor outer segments (POS). POS are degraded in lysosomes. This process is called heterophagy that should be differentiated from autophagy, although lysosomes are regulating both processes. Accumulated lipofuscin during AMD development disturbs both processes heterophagy and autophagy [14][43]. Oxidative stress is known to induce autophagic flux in RPE cells, but chronic oxidative stress seems to lead to increased lysosomal lipofuscin but decreased lysosomal activity and autophagy. While these processes play an important role in the homeostasis of RPE cells, their impairment can lead to detrimental accumulation of damaged organelles and abnormal or toxic proteins [7][10][36,39]. Disturbed proteostasis is clearly demonstrated by the accumulation of lysosomal lipofuscin in the RPE and extracellular drusen deposits between the RPE and choriocapillaris [6][35]. It is shown that lysosomal degradative functions can be further inhibited in RPE cells by a lipofuscin, and accumulation of lipofuscin in the RPE is a sign of cellular senescence in AMD. Intralysosomal accumulation of toxic lipofuscin can also sensitize lysosomes to visible light, oxidative stress, jeopardize lysosomal stability and lead to cellular apoptosis [15][44].
3.2. Role of Choriocapillaris in nAMD Development
It should be noted that drusens together with age and disease related Bruch’s membrane (BrM) thickening increase the barrier between the choriocapillaris and the highly metabolically active outer retina, resulting in critically reduced metabolite delivery (
Figure 23)
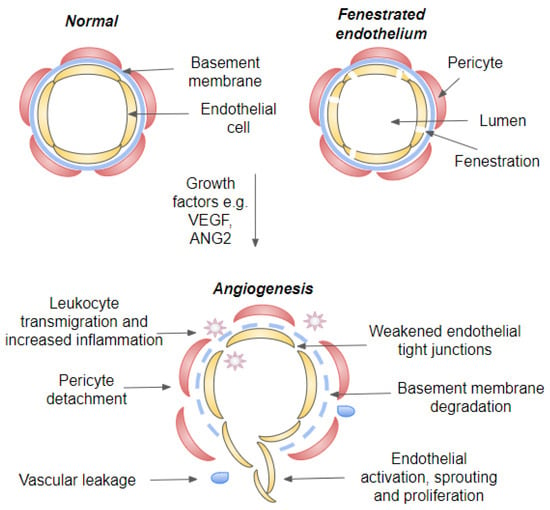
[1][16][1,54]. Choriocapillaries are the innermost region of choroid and are located just under the BrM and RPE. In addition, they have a rather large diameter (20–50 μm) and fenestrated endothelium to support the delivery of nutrients and export of waste products (
Figure 34). Under normal conditions, 85–90% of oxygen demand of outer retina is delivered by the choriocapillaris, while the remaining 10–15% is obtained from the deep retinal capillary plexus. For the entire retina, the choroid is responsible ~60 % of oxygen and ~75% nutrient supply. In the outer retina, the majority of oxygen is consumed in the photoreceptor inner segment where densely packed mitochondria produce ATP via aerobic respiration. To respond to the retina’s high rate of oxygen consumption, the choroid is perfused at a high rate and the drop in blood oxygen concentration along the vascular plexus to just 1% between arterioles and venules
[17][18][47,55]. Thus, the proper functioning of choriocapillaries is critical to support RPE and the rest of the retina. It is observed that during aging and early stages of AMD development, choroidal blood flow drops and both choroid and choriocapillaries become thinner. This degeneration includes loss of capillaries and reduction in endothelial cell fenestrations.
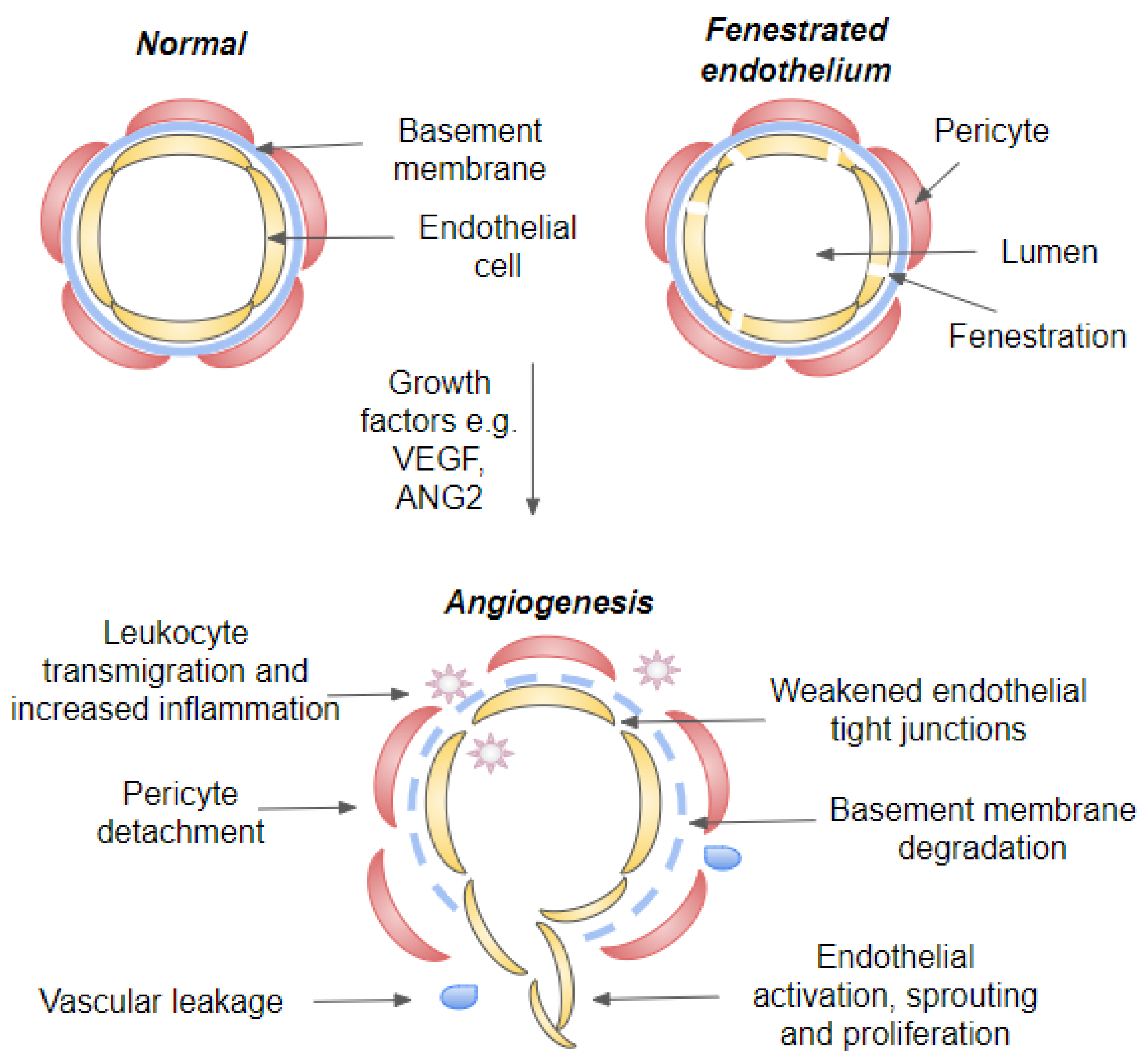
Figure 34. Illustration of a blood vessel in quiescent and angiogenic state. Cross-section of a capillary, fenestrated capillary and blood vessel during neovascularization. During vascular homeostasis, endothelial cells are connected by tight junctions and covered by both basement membrane and pericytes. Growth factors such as VEGF-A and ANG2 induce angiogenic switch that will lead to weakening of endothelial tight junctions, pericyte dropout and initiation of angiogenesis. Endothelial destabilization will further lead to vascular leakage and leukocyte transmigration. Abbreviations: VEGF, vascular endothelial growth factor; ANG, angiopoietin.
3.3. Senescence in AMD
During AMD progression, decreased autophagy is also associated with cellular senescence and senescence-associated secretory phenotype is associated with the release of ROS, selective growth factors, and inflammatory cytokines, chemokines and proteases. Senescent cells also promote the senescence of surrounding cells’ secretory phenotype, which can contribute to the maintenance of a chronic state of low-grade inflammation in tissues and organs. They are also apoptosis resistant, failing to enter programmed cell death and rather aggregate instead. Therefore, they may regulate drusen biogenesis and advanced GA and nAMD development [19][20][56,57]. Term immunosenescence is used to describe altered immune functions during aging.
3.4. Fibrosis in nAMD
The transition from early to advanced AMD shares many features with a defective wound healing response resulting from underlying oxidative stress, degeneration and chronic inflammation (
Figure 23). Subretinal fibrosis is a characteristic of the end-stage of AMD, resulting in a permanent vision loss. Wound healing will occur when injured tissue activates recruitment and activation of inflammatory cells and fibroblasts. In AMD, fibrosis is the product of defective and excessive wound healing response, which has unique characteristics as in nAMD it can originate from pre-existing neovascular membrane
[21][22][60,61]. Multiple cell types, including fibroblast, fibrocytes, macrophages, RPE cells and endothelial cells, may potentially participate to this process. Matrix-producing mesenchymal cells in subretinal fibrotic lesions can for example originate from the retinal pigment epithelium and/or choroidal endothelial cells through epithelial–mesenchymal transition (EMT,
Figure 23) and endothelial–mesenchymal transition (EndMT). Moreover, macrophages are able to transdifferentiate to myofibroblasts through macrophage-mesenchyman-transition (MMT). Majority of mesenchymal cells present in fibrotic lesions are myofibroblasts, which are not normally present in adult tissues
[21][22][60,61]. It is believed that pro-inflammatory cytokines can promote differentiation and activation of myofibroblasts (e.g., EndMT and EMT). RPE cells and infiltrating macrophages are believed to be a major source of these cytokines. During EMT, EndMT and MMT cells experience several biochemical and morphological changes, they, for example, become more motile and adaptable. They lose cell-to-cell contacts and cell polarity, and further disengage from their basal surface and basement membrane in order to migrate and produce extracellular matrix components. Fibrosis in nAMD is considered to originate from neovascular membranes and to be a consequence of a fibrovascular scarring resulting from inflammation and hypoxia-driven angiogenesis
[21][22][23][24][60,61,62,63]. Fibrosis may in some circumstances restore the protective barrier but can also progressively remodel and destroy normal tissue, leading to contracture and distortion of tissue architecture. As retinal visual function is built on highly organized anatomical layers and tightly coordinated cellular interactions, subretinal fibrosis will lead to a profound and often irreversible visual impairment. Currently, the pathogenetic mechanisms of subretinal fibrosis are poorly understood, and there is no therapy that would prevent excessive subretinal fibrosis
[22][61]. It is hypothesized that during nAMD progression, RPE cells might avoid cell death and escape from the stressful microenvironment and oxidative insult via EMT, but the development of fibrosis is also linked to angiogenesis
[20][21][25][57,60,64].
4. Angiogenesis and Inflammation
Blood vessel formation can be divided into vasculogenesis and angiogenesis. Vasculogenesis involves a de novo development of blood vessels and differentiations of endothelial cells from angioblasts and occurs mainly during embryogenesis. Whereas angiogenesis is a development of vascular capillaries from pre-existing blood vessels and is responsible for the further modeling of the vascular networks
[26][71]. It is a fundamental process during development and tissue regeneration. Angiogenesis is stimulated when hypoxic, diseased, or injured tissues produce and release factors that promote angiogenesis (
Figure 23). These proangiogenic growth factors stimulate the migration and proliferation of endothelial cells from preexisting vessels and, subsequently, the formation of capillary tubes and the recruitment of other cell types to generate and stabilize new blood vessels (
Figure 34)
[26][71].
Angiogenesis and inflammation are seemingly two different processes but are closely linked together as especially angiogenesis occurring in adult organisms is often linked to the inflammation
[27][66]. Inflammation is a cellular response to factors that challenge the homeostasis of cells or tissues and is intended to eliminate foreign or damaged material. At the beginning of an inflammatory response, foreign or damaged material becomes sensed by various pattern recognition receptors (PRRs). The ligand recognition process activates intracellular signaling pathways, resulting in the production of numerous proinflammatory mediators
[7][28][29][30][36,72,73,74]. Inflammation can destroy or inactivate invading pathogens, remove waste and debris, and permit restoration of normal function, through either resolution or repair. The goal of the inflammatory process is to repair damaged tissue in order to restore the typical tissue architecture, thus maintaining cellular/tissue homeostasis. After resolutions of inflammation, tissue structure should be normal, whereas repair leads to a functional, but morphologically altered organ. During acute inflammation, tissue damage is followed by resolution, whereas in chronic inflammation, damage and repair continue simultaneously. The initial inflammatory response is often acute, and depending on the circumstances, may evolve into chronic inflammation. Although inflammation is usually beneficial to the organism, it may also lead to tissue damage, resulting from the escalation of chronic inflammation
[7][28][29][30][36,72,73,74].
4.1. Inflammatory Signaling Cascades
A number of signals, ranging from microbes and other foreign material to mechanical tissue injury and autoantigens, can stimulate inflammation. Although it is a crucial survival mechanism, prolonged inflammation is detrimental and plays a role in numerous chronic age-related diseases
[27][31][66,78]. Inflamed tissues are characterized by a hypoxia and immune cell infiltration, a process that will result as an upregulation of molecular and cellular mechanisms that will regulate angiogenesis (
Figure 23 and
Figure 34). Inflammation-associated angiogenesis is linked to several pathophysiological processes, such as cancer and scar formation. During acute inflammation, fluid and immune cells accumulate at the site of injury due to changes in small blood vessel integrity. Cells, which are damaged by, e.g., cellular stress or infectious agents, expose molecules called as alarmins or damage associated molecular patterns (DAMP)
[7][29][36,73]. These molecules become sensed by a variety of cells that express PRRs, which will induce amplification of immune response. During this process, inflammasome and nuclear factor kappa B (NF-κB) signaling pathways become activated and a number of proinflammatory cytokines and other inflammatory mediator will be released. The cytokines and inflammatory mediators include molecules such as VEGF, IL-1α, IL-1β, IL-8 and TNF-α, as well as histidine, thrombin and fibrinogen. These molecules activate endothelial cells, induce vasodilation and increase vascular permeability, which will further facilitate immune cell transmigration to eliminate the aggressive agent
[7][29][36,73]. Endothelial cell activation is characterized by increased expression of leukocyte adhesion molecules, cytokines, growth factors, HLA molecules and will lead to changes in endothelial cell junctions and surrounding pericytes (
Figure 34). Vascular phenotype will further change from antithrombotic to prothrombotic, in order to prevent the spreading of a potential pathogen and platelets participate in the coagulation process in order to prevent blood loss from damaged vessels. Subsequently, vasodilation occurs, and permeability of blood vessels increases, allowing inflammatory mediators and immune response cells, including leukocytes and monocytes/macrophages, to infiltrate damaged tissue. Which will further modify the microenvironment in the retina
[29][31][32][33][70,73,78,79].
4.2. Blood-Retinal-Barriers
Macular edema results due to vascular hyperpermeability and is identified by swelling of the central portion of the human retina, and is associated with increased retinal thickness. It can be defined as an excess of fluid within the retinal tissue (
Figure 23). Interstitial spaces of the retina are normally relatively dry as free leakage of fluid and protein from the macular vasculature is prevented by the blood-retinal-barrier (BRB). BRB can be divided into outer BRB (oBRB) and inner BRB (iBRB), which control the passage of substances in outer and inner retina, respectively. Outer BRB includes the choroid, Bruch`s membrane and RPE
[34][81]. Vasculature of oBRB includes choriocapillaries, which are maintained by the VEGF produced from the basolateral side of the RPE cells and actively supply nutrients as well as remove waste products from outer retinal layers
[32][35][70,82]. As choriocapillaries are fenestrated, they do not provide barrier by themself, but barrier is formed by Bruch`s membrane and RPE
[32][34][70,81]. Interestingly, ultrastructure of choriocapillaries is further polarized as fenestrations locate on the retinal side of the vessels and pericytes are found from the outer wall facing sclera. This structure further facilitates efficient movement of macromolecules between retina and choroid
[36][83]. Paradoxically, fenestrated vasculature is dependent on constant VEGF supply
[37][84], but choriocapillaries in human still seem to tolerate long-term treatment with anti-VEGF inhibitors in ocular conditions. As a comparison, the kidneys have also fenestrated vasculature, and it has been reported that cancer treatment with anti-VEGF treatments will cause changes in kidney function
[38][39][40][85,86,87]. Furthermore, it has been demonstrated by mouse models, that maintenance of horiocapillaris is dependent on constant VEGF supply
[35][37][82,84].
4.3. Inflammatory Cell Recruitment
Already 1985 Penfold et al., described the involvement of immunocompetent cells in early, intermediate and late stage of AMD. They suggested that macrophages, fibroblast, lymphocytes and mast cells play a role in neovascularization, atrophy of RPE and Bruch`s membrane breakdown
[41][91]. During the inflammation process, leukocytes sense the chemokine gradient originating from the inflamed tissue and begin to make contact with the adhesion molecules expressed by endothelial cells to permit their tighter binding to the vascular endothelium. Finally, leukocytes leave the circulation by following the chemokine gradient and move towards the damaged tissue, where they become activated
[28][72]. Studies done with an inducible model of photoreceptor death in mice showed infiltration of 12 distinct subpopulations of microglia, monocytes and macrophages
[42][92]. Previously mentioned study did not identify markers for neutrophils, but infiltration of neutrophils has been described from early AMD patient samples and studies done with mouse models suggested the role of neutrophils in retinal degenerations
[42][43][92,93].
The role of the immune system during acute tissue damage and defense against foreign antigens has been well characterized, but its role for development of chronic age-related conditions should be more deeply understood. During the development of AMD, aging and oxidative stress will lead to the accumulation of waste product, which will endure para-inflammation to repair and remodel tissue through activation of microglial, macrophage and complement system
[7][44][36,95]. Neuroinflammation is always accompanied by microglial activation with the release of inflammatory mediators and phagocytosis. Microglia play a role both in innate as well as adaptive immunity and are resident cells in central nervous system. Under physiological conditions, they are in inactive state and release neuroprotective as well as anti-inflammatory factors. Instead, when damage occurs in the central nervous system, they can mediate both protective and harmful actions. Beneficial actions limit the further injury and include removal of waste and degenerated cells as well as the secretion of neuronal survival factors. However, microglia can also promote persistent inflammation and recruit additional inflammatory cells
[7][44][36,95]. It has been shown that during AMD, migroglial accumulates in the subretinal spaces at the sites of choroidal neovascularization and retinal degeneration.
During inflammation, the arrival of increasing numbers of immune cells exacerbates the inflammatory response and induces chronic inflammation. It is not only white blood cells that enter the inflamed tissue but also fluids and various plasma proteins gain access to the damaged tissue (
Figure 23 and
Figure 34). Extravasated plasma proteins such as fibrinogen may further stimulate neovascularization. Inflammatory cells, including macrophages, lymphocytes, mast cells, and fibroblasts, and the angiogenic factors they produce, can also stimulate vessel growth
[7][28][36,72]. Many proinflammatory cytokines, such as tumor necrosis factor (TNF)-α, IL-6, IL-1β, IL-8 and VEGF, have both angiogenic and proinflammatory activity
[45][46][47][48][49][14,105,106,107,108].
4.4. Effects of Hypoxia
Inflammation can promote angiogenesis in several ways, e.g., inflammatory tissue is often hypoxic, and hypoxia can induce angiogenesis through upregulation of factors such as VEGF
[28][29][72,73]. Normally, mammalian cells are dependent on the oxygen and nutrients so they locate within 100 to 200 μm of blood vessels
[50][114], but if tissue environment faces changes e.g., due to inflammation induced swelling, the existing blood vessels may not be able to provide enough oxygen to the tissue and the hypoxia may occur. Interestingly, hypoxia is one of the factors that can be regarded as a common nominator for both inflammation and angiogenesis, while reduced choroidal perfusion and hypoxia have been also linked to the development of the AMD (
Figure 23).