Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Yu Zakharova | -- | 3357 | 2023-01-31 18:18:58 | | | |
| 2 | Conner Chen | + 28 word(s) | 3385 | 2023-02-01 02:15:30 | | | | |
| 3 | Conner Chen | + 7 word(s) | 3392 | 2023-02-02 09:43:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ishina, I.A.; Zakharova, M.Y.; Kurbatskaia, I.N.; Mamedov, A.E.; Belogurov, A.A.; Gabibov, A.G. Major Histocompatibility Complex Class II Presentation in Autoimmunity. Encyclopedia. Available online: https://encyclopedia.pub/entry/40682 (accessed on 08 August 2026).
Ishina IA, Zakharova MY, Kurbatskaia IN, Mamedov AE, Belogurov AA, Gabibov AG. Major Histocompatibility Complex Class II Presentation in Autoimmunity. Encyclopedia. Available at: https://encyclopedia.pub/entry/40682. Accessed August 08, 2026.
Ishina, Irina A., Maria Y. Zakharova, Inna N. Kurbatskaia, Azad E. Mamedov, Alexey A. Belogurov, Alexander G. Gabibov. "Major Histocompatibility Complex Class II Presentation in Autoimmunity" Encyclopedia, https://encyclopedia.pub/entry/40682 (accessed August 08, 2026).
Ishina, I.A., Zakharova, M.Y., Kurbatskaia, I.N., Mamedov, A.E., Belogurov, A.A., & Gabibov, A.G. (2023, January 31). Major Histocompatibility Complex Class II Presentation in Autoimmunity. In Encyclopedia. https://encyclopedia.pub/entry/40682
Ishina, Irina A., et al. "Major Histocompatibility Complex Class II Presentation in Autoimmunity." Encyclopedia. Web. 31 January, 2023.
Copy Citation
Antigen presentation by major histocompatibility complex class II (MHC-II) molecules is crucial for eliciting an efficient immune response by CD4+ T cells and maintaining self-antigen tolerance. Some MHC-II alleles are known to be positively or negatively associated with the risk of the development of different autoimmune diseases (ADs), including those characterized by the emergence of autoreactive T cells. Apparently, the MHC-II presentation of self-antigens contributes to the autoimmune T cell response, initiated through a breakdown of central tolerance to self-antigens in the thymus.
autoimmune diseases
autoreactive T cells
human leukocyte antigen
1. MHC-II Presentation of Low-Affinity Self-Antigens
Major histocompatibility complex class II (MHC-II) molecules are synthesized in the endoplasmic reticulum (ER); the peptide-binding groove is occupied by the invariant chain Ii (CD74 fragment) to prevent degradation and premature binding of self-antigens by the complex. As they migrate to the late endosomal compartments with an acidic environment, proteolytic enzymes shorten the invariant chain to a shorter class II-associated invariant chain peptide (CLIP) [1]. Protein antigens are proteolytically processed in endosomes as well (Figure 1). The peptide-binding groove of an MHC class II molecule has nine pockets that can accommodate certain amino acid residues of the peptide, typically stabilized by noncovalent bonds [2]. The P1, P4, P6, and P9 anchor residues interact with the groove of MHC-II, thus forming a binding register, while the remaining residues of the peptide are oriented in the opposite direction for T cell receptor (TCR) binding. Presumably, if there is a shift in the binding register between antigenic peptides and MHC molecules, the peptide–MHC-II complex (pMHC) complex can interact with an entirely different TCR.

Figure 1. MHC-II maturation and antigenic peptide loading. MHC-II molecules are synthesized in the endoplasmic reticulum (ER) and loaded with an invariant chain (Ii). The MHC-II complex with Ii is transported through the Golgi to the late endosome. Endosomal proteases process antigens to short peptides and Ii to shorter class II-associated invariant chain peptide (CLIP). The binding of a nonclassical HLA-DM (DM) molecule to MHC-II promotes CLIP exchange to the antigenic peptide with an optimal binding register. The formed pMHC complex is transported to the surface of the antigen-presenting cell (APC) for CD4+ T cell recognition.
The nonclassical MHC molecules HLA-DM (DM) and HLA-DO (DO) in humans and H2-DM and H2-DO in mice play an important role in exchanging CLIP for endosomal antigens [3]. By interacting with MHC-II, DM has the “editing” function: it ensures the binding of peptides with higher affinity for the MHC molecule compared with CLIP, as well as increases the loading/dissociation rate of the antigen, but it causes no effect on the equilibrium affinity [4][5]. Apparently, DM facilitates the presentation of antigen epitopes with the optimal binding register on MHC-II [6][7]. DO also performs an editing action, interacting with DM and regulating its catalytic function by preventing its binding to MHC-II [8]. Comparing MHC-II immunopeptidomes from two cell lines, DO knockout or not, it was shown that only the DR1+, DM+, and DO+ lymphoblastoid cell lines presented specific antigens on MHC-II [9]. Thus, DO contributes to the diversification of the MHC-II-presented antigenic repertoire. In experiments with DM-knockout mice, CLIP was not efficiently exchanged with other peptides, thus limiting the MHC-II antigen diversity and causing incomplete negative selection of CD4+ T cells in the thymus [10]. The repertoire of antigens presented by MHC-II can vary significantly depending on the DM/DO ratio in the cell [11]. Presumably, DM “editing” decreases the number of antigens, characterized by a low affinity for MHC-II, on the antigen-presenting cell (APC) surface, as was shown for DQ1 and DQ6 [12], as well as for DR3 alleles [4]. Analysis of the immunopeptidome of thymic antigen-presenting cells (APCs) revealed that most antigens detected on MHC-II have a high affinity for MHC-II molecules [13][14]. Most likely, the effect of nonclassical MHC molecules is one of the important factors ensuring the highly competitive conditions for MHC-II ligand binding, which eventually results in the presentation of higher-affinity antigens.
It should be emphasized that the presentation of preferentially high-affinity antigens in the thymus may have its own shortcomings. It has been demonstrated that the MHC-II-presented antigen repertoire from tissues affected by autoimmune processes mostly consists of low-affinity peptides [15][16]. The abundance of self-antigens increases in peripheral tissues (e.g., myelin basic protein (MBP) in the central nervous system (CNS) [17][18][19] and insulin in the pancreas [20]). These protein self-antigens can be processed in the extracellular environment outside APCs. Next, the resulting antigenic fragments, characterized by a low affinity for MHC-II molecules, can be loaded directly on the APC surface or can enter early endosomes with low DM levels, thus escaping DM editing, and as a result, can be presented on the cell surface (Figure 2A,B). Certain MHC-II risk alleles bind CLIP with diminished affinity, which additionally promotes the binding of low-affinity antigenic peptides [21][22]. Rapid dissociation of CLIP enables the generation of empty complexes without DM involvement, which may potentially bind low-affinity antigenic peptides. Due to the spontaneous release of CLIP, low-affinity antigenic peptides are able to bind MHC-II in early endosomes without DM assistance or directly on the cell surface. Therefore, pMHC complexes on the APC surface at the periphery can activate T cell receptors (TCRs) that have not undergone negative selection due to the low abundance of this pMHC in the thymus. For multiple sclerosis (MS) patients, there was detected an immune response to full-length MS autoantigen proteolipid protein (PLP), naturally processed by APCs, with 2 immunodominant epitopes generation. Alternatively, the T cell response to several PLP synthetic fragments with high affinity to MS-associated MHC-II alleles was also observed after additional T cell stimulation by these peptides [23]. Since these fragments are normally hidden in a protein fold and are inaccessible for the endosomal proteases, the T cell activation seems to be the result of extracellular PLP processing and loading on MHC-II. Thus, the classical intracellular processing of antigen involves its capture by the APC with proteolytic processing and generation of fragments with optimal binding registers with the participation of HLA-DM in late endosomes, followed by subsequent exposure of pMHCs on the APC surface. These pMHCs take part in the negative selection process in the thymus, induce T cell clonal deletion and, after all, no autoreactive T cell response is observed at the periphery. Since pMHCs with “suboptimal” fragments were present in the thymus at low levels, autoreactive T cells might engage them at the periphery due to the failure of negative selection in the thymus.
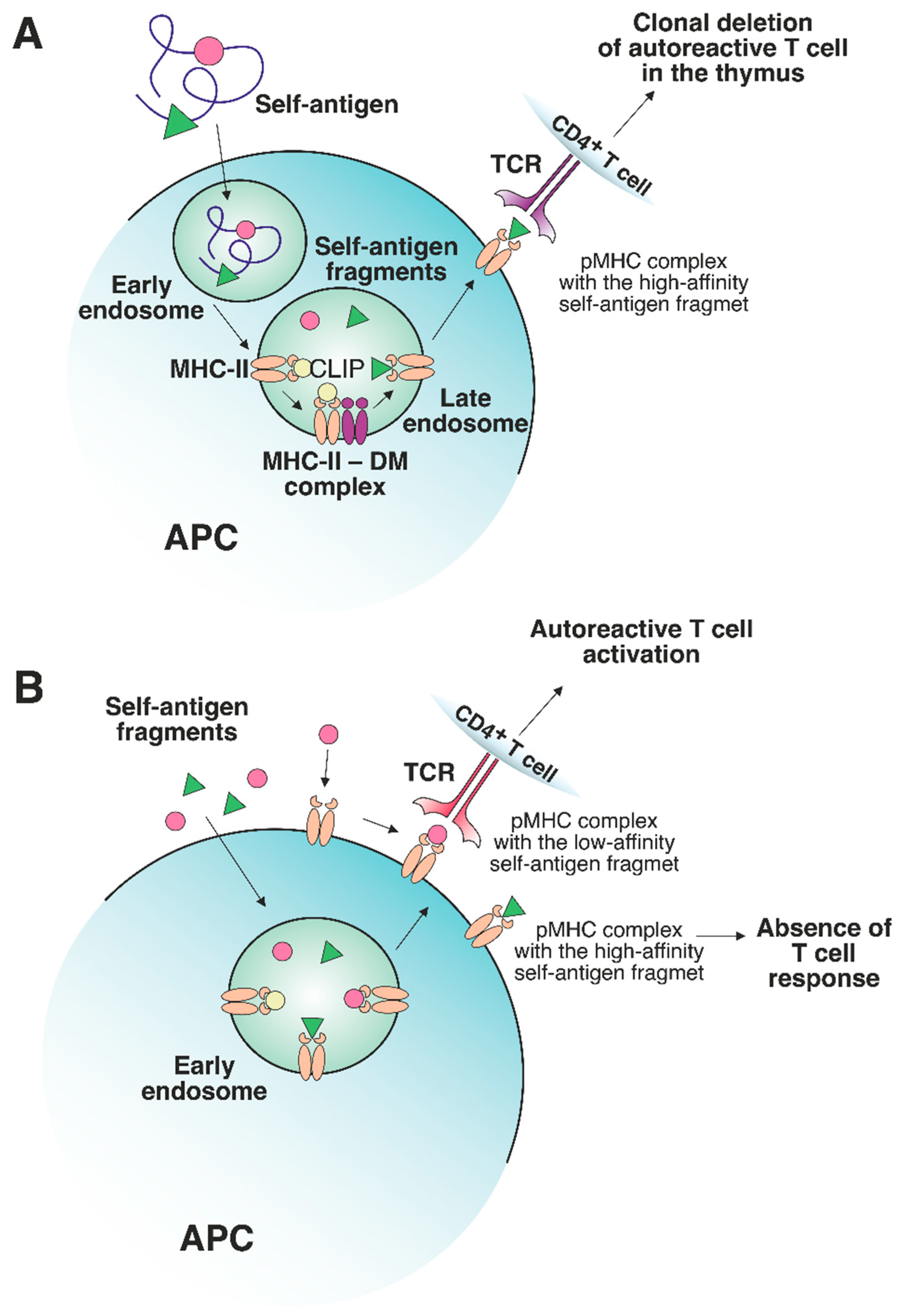
Figure 2. The role of a nonclassical DM molecule in the editing of the self-antigen repertoire presented by MHC-II. (A) In a thymic APC, the self-antigen is processed through a classical pathway under exposure to cellular proteolytic enzymes. Next, CLIP is exchanged to the high-affinity fragment in late endosomes under DM control. The stable pMHC complex is then transferred to the APC surface, where the T cells specific to this pMHC undergo clonal deletion. (B) If an excessive amount of certain self-antigens, processed outside APCs, is present in peripheral tissues, then these self-antigen fragments can be loaded on MHC-II molecules in early endosomes or on the surface of APCs without the recruitment of DM. The absence of DM editing allows the emergence of either stable or unstable pMHC complexes. The pMHCs, containing high-affinity fragments, do not elicit a T cell response due to the deletion of autoreactive T cells in the thymus. Unstable complexes with low-affinity fragments can bind autoreactive T cells since such T cells are not susceptible to clonal deletion.
2. The Peculiarities of the Interaction between TCRs and Self-pMHC Complexes: Links with Autoimmunity
Generally, CD4+ T cell TCRs recognize foreign antigens within MHC-II molecules [24][25][26] with CD4 receptors playing the key role in the induction of the T cell signaling cascade [27][28][29]. The analysis of trimolecular complexes between TCRs, MHC-II, and exogenous antigens has revealed some structural similarity in the TCR binding topology [30]. The TCR is arranged diagonally with respect to the antigen residing in the peptide-binding groove limited by α-helices. The variable regions of α and β TCR chains interact with the β and α chains of the MHC-II molecule, respectively [31]. The most structurally diverse CDR3 fragments of α and β TCR chains are located above the P5 residue of the bound peptide, while the CDR1 and CDR2 fragments interact with the α-helices of MHC-II molecules. The binding of TCR HA1.7 to the fragment of hemagglutinin HA protein within DR1 is an example of canonical interaction [32].
It was shown that, by contrast with TCRs binding bacterial or viral antigens on MHC-II, some autoreactive TCRs have an alternative binding topology. Thus, the TCR from an MS patient (Ob.1A12) specific for the fragment of myelin basic protein (MBP), which is presented on the HLA-DR2b risk allele molecule, interacts mainly with the N-terminus of the MBP fragment [33]. Furthermore, this TCR does not reside in the canonical diagonal position: its orientation angle is 110°, as opposed to 70° for HA1.7. The main interaction with the pMHC complex occurs due to the binding of the CDR3 region of TCR with the P2 residue of the MBP peptide fragment. Other TCRs, Ob.2F3 and Ob.3D1, obtained from the same donor with MS, also have an alternative binding topology, as examined using computational simulation [34]. Another TCR, 3A6, binds the MBP fragment in the complex with different DR15 allomorph DR2a, which is positively associated with the risk of developing MS. The 3A6 also has a suboptimal topology and low affinity for pMHC, similar to other autoreactive TCRs [35]. Its CDRs are shifted toward the N-terminus of the antigen, and CDR3 is also located above P2 of the MBP fragment. Nevertheless, similar to HA1.7, 3A6 binds diagonally with respect to pMHC. Therefore, Ob.1A12 and 3A6 have low affinity for pMHC, and their binding topology is alternate to anti-viral and anti-bacterial TCRs, which may potentially facilitate the escape from negative selection mechanisms in the thymus. Low-affinity TCRs were also reported in the pathogenesis of type 1 diabetes (T1D). The repertoire of the autoreactive T cells, infiltrating pancreatic islets, exhibited low self-reactivity and promoted the development of T1D [36]. Presumably, the interaction between these T1D TCRs and self-antigen-carrying pMHCs is also characterized by alternate topology. The MBP fragment presented by DQ1 is recognized by the Hy.1B11 TCR with relatively high affinity [37]. These TCRs have canonical localization but are significantly shifted with respect to the DQ1α chain so that their interaction with the antigen is substantially limited. Only the CDR3 region of the Hy.1B11α chain contacts the MBP fragment. Presumably, the unusual topology of autoreactive TCRs binding to pMHC impedes activation by the CD4 molecule and, therefore, clonal deletion of T cells at the negative selection stage. This may explain the existence of T cells with similar TCRs (such as Hy.1B11) in the periphery [38]. The unusual topology of the trimolecular complex often implies the participation of a low-affinity TCR. However, autoimmune TCRs with a high affinity for pMHCs have also been reported. It is known that DQ molecules have a lower expression level than DR molecules. Therefore, appropriate autoreactive TCRs might require higher affinities for DQ pMHC complexes to overcome clonal deletion in the thymus [37]. Another possible explanation of high-affinity autoreactive TCR emergence at the periphery is that the autoantigenic fragment may be a weak binder in the pMHC complex. As previously discussed, pMHC carrying low-affinity peptides allows T cells to escape negative selection. Nevertheless, they can further bind autoreactive TCRs at the periphery. Interestingly, the weak interaction between the MBP fragment and the product of the risk allele DR4 is stabilized by the MS2-3C8 TCR involved in canonical high-affinity interaction with pMHC [39]. The structures of the trimolecular complexes of three autoreactive TCRs in model mice with experimental autoimmune encephalomyelitis (EAE), which bind the MBP fragment in complex with I-Au, have revealed canonical binding.
3. The Effect of MHC-II-Presented Self-Antigen Modifications on Autoreactive TCR Recognition
Antigens processed for further presentation by MHC-II molecules can undergo various posttranslational modifications (PTMs), including glycosylation, iodination, citrullination, etc. (Figure 3). PTMs can either occur spontaneously or be induced by various enzymes. Autoreactive T cell responses to modified self-antigens have been observed for many autoimmune diseases (ADs). Supposedly, the modified antigens (neoantigens) are either totally not present in the thymus or their quantity is extremely low. These modifications presumably take place in peripheral tissues and are absent when the antigen is presented by medulla thymic epithelial cell (mTEC) cells in the thymus, which has been demonstrated for glycosylated type II collagen for the development of rheumatoid arthritis (RA) [40]. Although modified antigens are available in the thymus due to the presence of migratory APCs, their quantity is probably insufficient for negative T cell selection in the thymus. Thus, thyroglobulin, which is the autoantigen in patients with Hashimoto’s thyroiditis, is present in the thymus in its nonmodified form, whereas the level of its iodinated form, which seems to initiate the autoreactive T cell response, is extremely low in the thymus and is insufficient for central tolerance to be established [41].
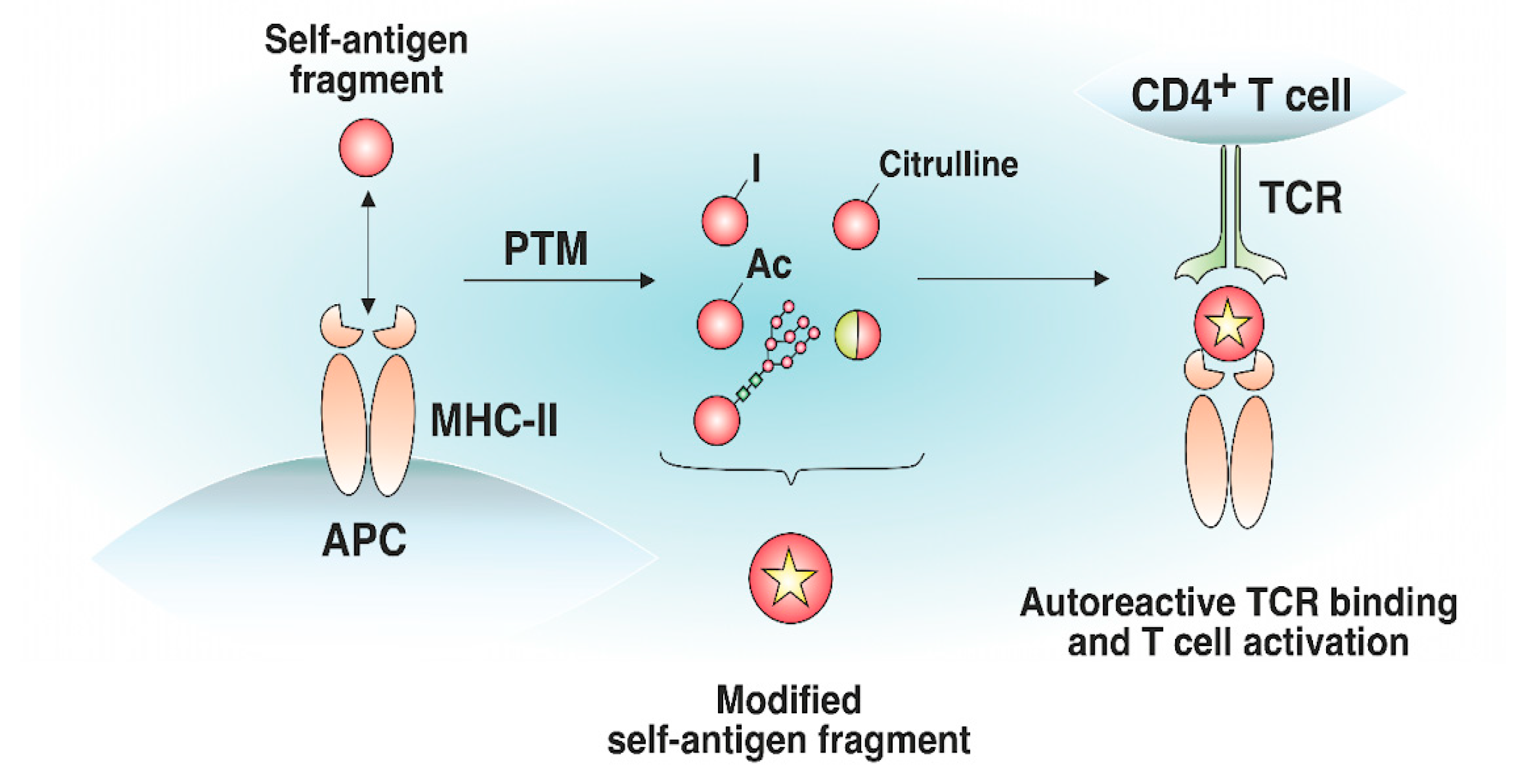
Figure 3. Effect of posttranslational modifications of self-antigens on trimolecular complex assembly. Certain self-antigen fragments do not bind to MHC-II molecules, or their binding affinity is low. Similar self-antigen with posttranslational modifications (iodination, acetylation, glycosylation, citrullination, generation of hybrid peptides, etc.) exhibiting increased affinity for MHC-II molecules, can appear in peripheral tissues. The resulting pMHC complexes can bind autoreactive T cells that have not undergone negative selection in the thymus.
Citrullination of antigens is described for a number of ADs, especially for RA; the positive charge of arginine is lost due to the conversion to citrulline, which ultimately alters the epitope affinity to MHC-II and TCR [42]. The products of risk MHC-II alleles for RA carry a consensus amino acid sequence in the antigen-binding groove, forming a positively charged P4 pocket–shared epitope (SE), which binds the polar amino acid residues of peptides at the P4 position (e.g., citrulline) and induces the activation of autoreactive T cells [43][44]. In addition to hosting a polar-neutral citrulline residue, SE can itself serve as the main contact zone for autoreactive TCRs and represent citrullinated fragments of the fibrinogen autoantigen that directly interact with autoreactive TCRs via the P2-citrulline residue [45]. Autoreactive T cells specific for citrullinated tenascin-C antigenic peptides, presented by DR4 molecules, were revealed in RA patients [46]. Additionally, glycosylation was reported to be responsible for the pathogenesis of RA. Analysis of the crystal structure of the trimolecular complex of an autoreactive TCR and type II collagen (Col2) neoantigen presented on DR4 has shown that lysine residues subjected to galactosylation are the key sites for TCR recognition [47]. Furthermore, the autoreactive T cell response is dependent on the galactosylation of lysine in the Col2259–273 fragment at position 264. Modified autoantigenic peptides also participate in T1D development. Several citrullinated and transglutaminated GAD65 epitopes are recognized by autoreactive CD4+ T cells in T1D preferentially to their unmodified form [48]. Autoreactive T cells specific for citrullinated glucokinase epitopes are linked to T1D pathogenesis [49]. Another example of the modified antigens impacting the pathogenesis of T1D is the generation of a disulfide bond. T cells recognize insulin fragments presented on DR4 molecules in T1D upon the assembly of a disulfide bridge between neighboring cysteine residues [50]. The citrullinated fragments of MBP and myelin oligodendrocyte glycoprotein (MOG) cause EAE due to the activation of autoreactive T cells [51][52][53]. In EAE, acetylation of the MBP fragment elicits an encephalitogenic T cell response [54][55][56].
4. The Effect of the Proinflammatory Environment on MHC-II Antigen Presentation and Autoreactive T Cell Engagement
The generation and expansion of pathological T cells may be attributed to the increase in MHC-II expression levels due to the proinflammatory environment induced by viral or bacterial infections. The impacts of viral and bacterial infections on antigen presentation are the proposed triggers of pathological CD4+ T cell expansion in ADs such as MS and T1D [57][58][59][60][61]. Earlier, a proinflammatory environment was implicated in TCR-independent bystander activation in different ADs [62][63]. Proinflammatory cytokine release and bacterial antigen presence trigger the elevated synthesis of MHC-II molecules to maximize the presentation of foreign antigens for an efficient CD4+ T cell response. High IFN-γ levels increase the number of MHC-II molecules on professional and nonprofessional APCs, thereby altering the composition of the MHC peptidome in the inflammatory area [64]. Additionally, APCs, activated in pathogenic conditions, express an increased number of costimulatory molecules [65][66]. Therefore, rare self-antigen pMHC, which cannot induce an autoreactive T cell response under normal conditions, might engage autoreactive TCRs in a proinflammatory environment (Figure 4). Concluding, cytokines induced by the inflammatory environment may potentially activate antigen-specific autoreactive T cells.
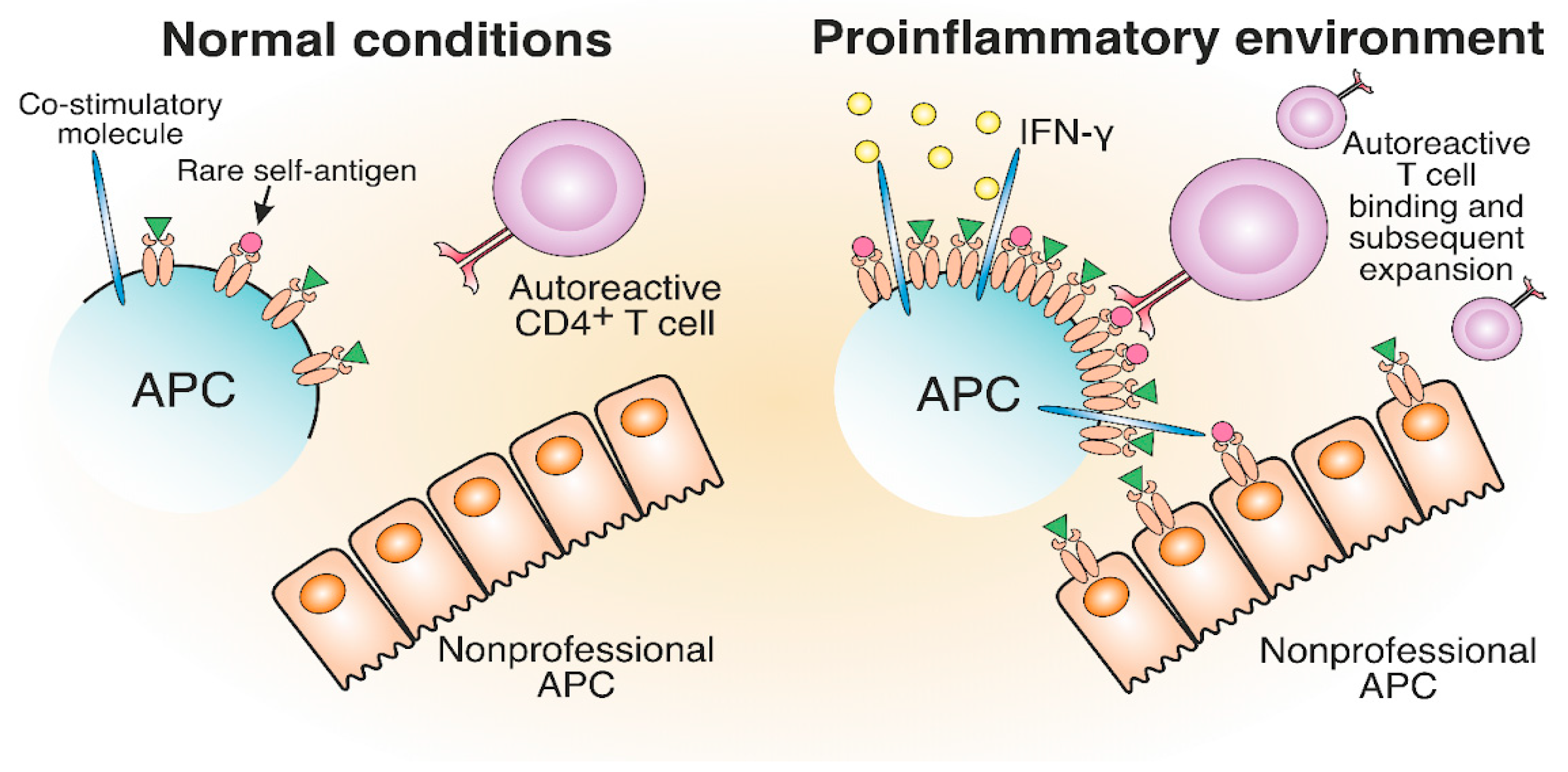
Figure 4. The development of autoreactive CD4+ T cells in a proinflammatory environment. The proinflammatory environment caused by viral or bacterial infection entails IFN-γ production, which results in increased MHC-II and costimulatory molecule expression compared to normal conditions. Therefore, rare self-antigens are present in a greater number, which can lead to the potential activation of autoreactive T cells.
A correlation between the inflammatory milieu and elevated MHC-II levels has been observed for several ADs. It was shown that MHC-II transcripts were upregulated by proinflammatory cytokine expression in β-cells of T1D patients [67][68]. IFN-γ induces the expression of I-Ag7 molecules on beta cells of non-obese diabetic (NOD) mice, leading to the emergence of CD4+ autoreactive T cells and the promotion of T1D [69]. In addition, IFN-γ promotes the expression of MHC-II on islet endothelial cells in NOD mice [70]. The transcription factors responsible for the expression of MHC-II are elevated in MS lesions of human brain tissue [71]. Aberrant MHC-II expression was also found on oligodendrocytes in EAE mice and MS patients [72]. The expression of MHC-II in mouse joints led to the development of severe erosive inflammatory polyarthritis [73]. Additionally, the inflammatory environment promotes the expression of MHC-II molecules on hepatocytes during autoimmune hepatitis and pMHC transfer via trogocytosis to interacting CD4+ T cells, which further amplifies the autoimmune response [74].
5. Molecular Mimicry between Self- and Foreign MHC-II Antigens May Lead to T Cell-Mediated Autoimmunity
Molecular mimicry refers to a structural similarity between self and exogenous antigens and potentially leads to autoimmunity [75][76][77]. The structural resemblance of antigens might result in the occasional misactivation of T cells (Figure 5). Autoreactive T cells from MS patients specific for MBP can cross-react with epstein–barr virus (EBV) nuclear antigen 1 (EBVNA1) [78][79]. Apparently, the virus-specific TCR was able to bind structurally similar self-antigen fragments presented on MHC-II and initiate an autoreactive T cell response. The probability of MS development is significantly increased when EBV infection is combined with the MHC-II risk allele HLA-DRB1*15:01. It was shown that CD4+ T cells specific for DR15-presented EBV antigens could cross-react with MBP [80].
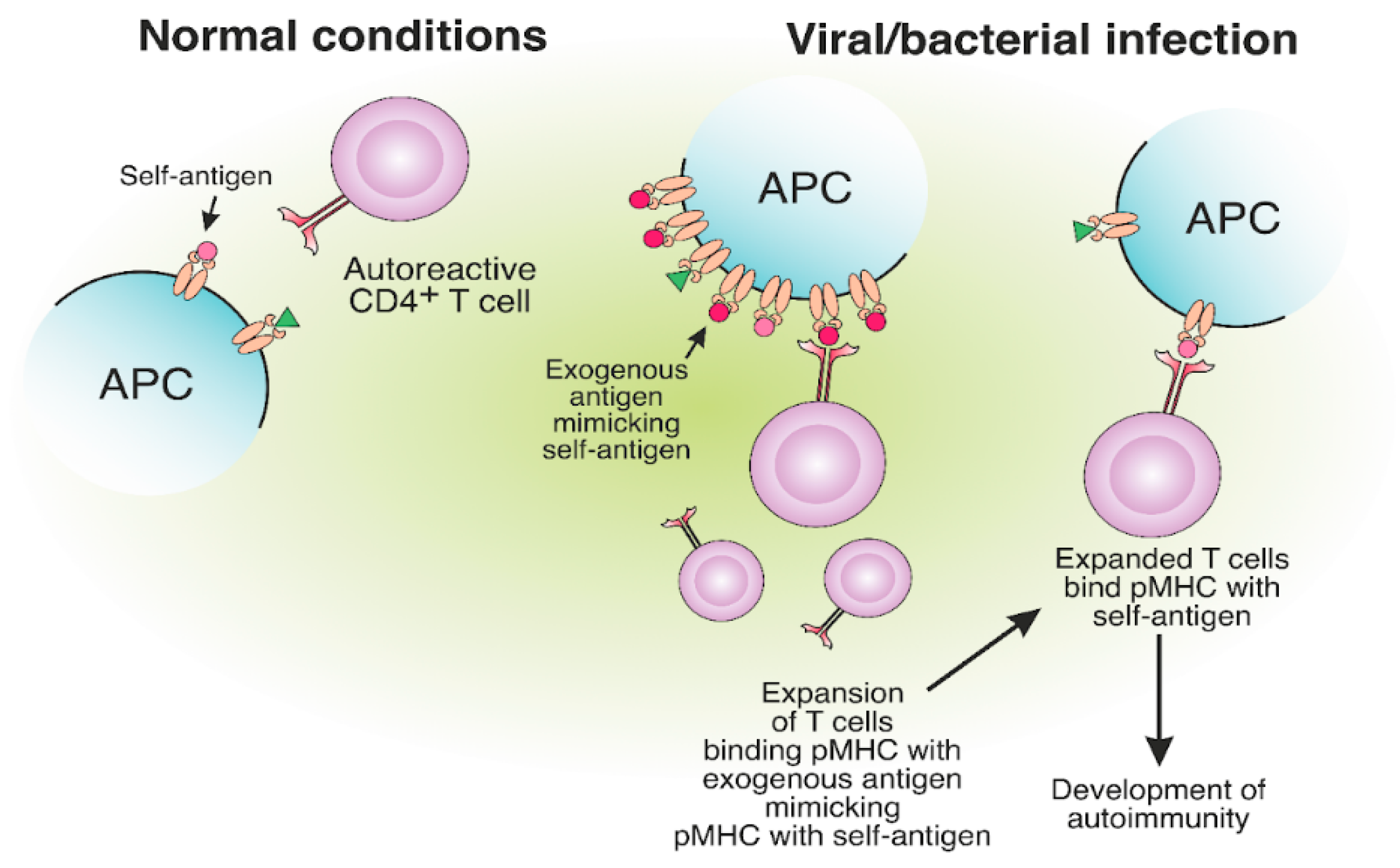
Figure 5. The expansion of autoreactive CD4+ T cells due to the molecular mimicry between foreign and self-antigens. The presentation of viral or bacterial antigenic peptides on MHC-II molecules, structurally similar to self-antigens, may result in the expansion of CD4+ T cells, specific to exogenous pMHC. Expanded CD4+ T cells, specific for foreign peptides, can bind the self-antigen pMHC, which results in the development of autoimmunity.
Narcolepsy is a rare autoimmune chronic neurological disorder positively associated with the HLA-DQB1*06:02 allele and characterized by targeting hypocretin neurons [81]. Cross-reactive T cells for hypocretin autoantigen (HCRTNH2) and flu HA (H1N1 2009 strain) antigens were shown to influence narcolepsy development [82]. T cell cross-reactivity between viral epitope neuraminidase (NA175–189) and self-epitope protein-O-mannosyltransferase 1 (POMT1675–689) is also involved in narcolepsy pathogenesis [83].
6. The Role of Protective MHC-II Alleles in the Development of Autoimmune Diseases
Certain MHC alleles provide so-called “protection” toward the development of immune-related diseases due to peculiarities of antigen presentation, which affect the engagement of T cells [84][85]. “Protective” allele means that the risk of AD initiation in individuals carrying this allele is statistically lower than that for healthy donors, representing the average population [86][87][88][89]. Interestingly, the protein products of MHC-II protective alleles often differ from the products of risk alleles only by several amino acid residues localized in the TCR contact sites or near the key positions of the peptide-binding groove [90]. Supposedly, the protective effect can be ensured due to the deletion of autoreactive T cells in the thymus and/or induction of Treg cells (Figure 6A).
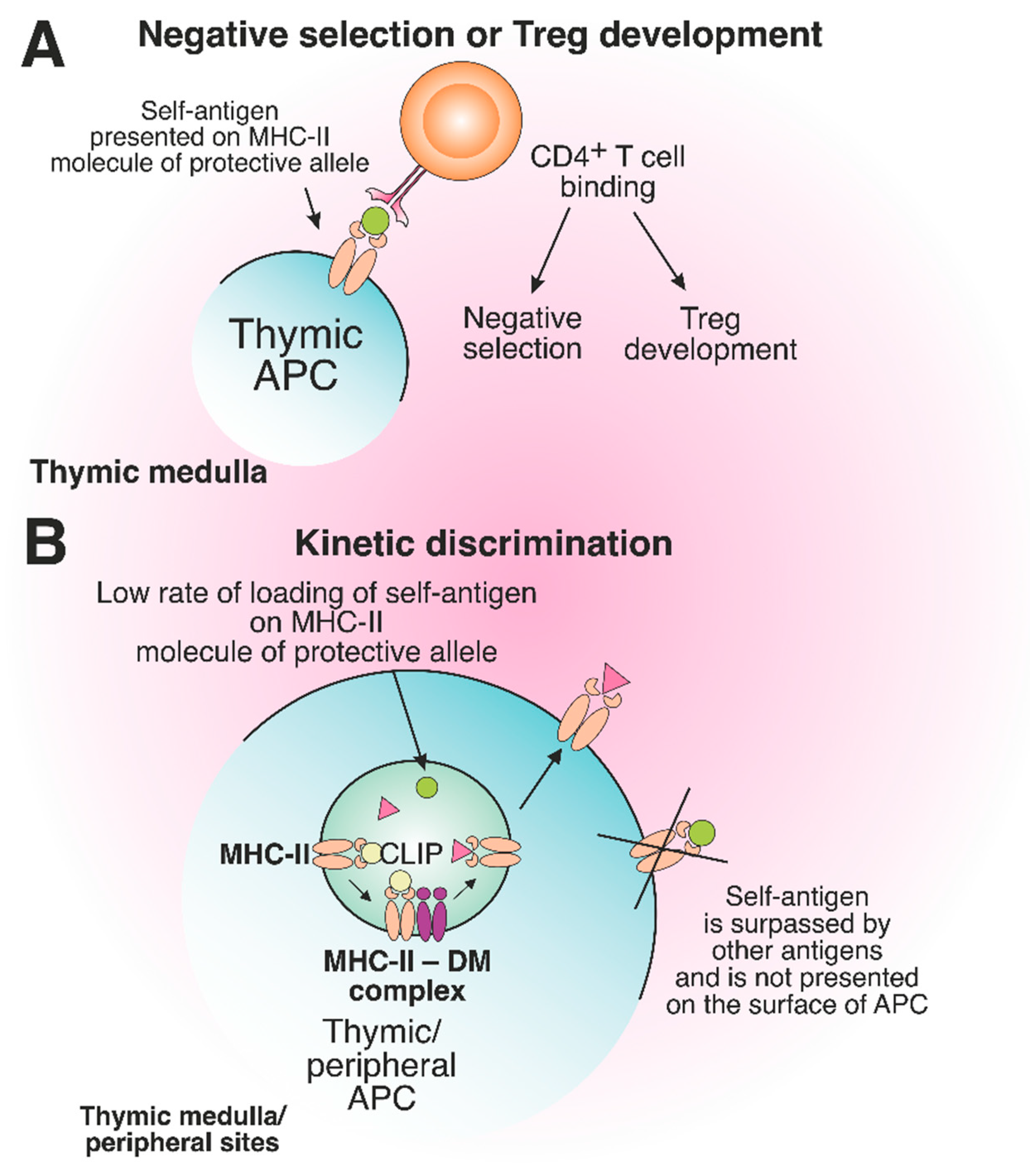
Figure 6. The role of protective MHC-II molecules in the suppression of autoreactive T cell development. (A) Recognition of the self-antigen presented on the product of the protective allele by CD4+ T cell results in negative selection or Treg development in the thymus. (B) The slow loading rate of self-antigen on the MHC-II molecule of the protective allele results in the failure of pMHC complex assembly on the surface of thymic or peripheral APCs.
References
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of Antigen Processing. Annu Rev. Immunol. 2013, 31, 443–473.
- Ishina, I.A.; Filimonova, I.N.; Zakharova, M.Y.; Ovchinnikova, L.A.; Mamedov, A.E.; Lomakin, Y.A.; Belogurov, A.A. Exhaustive Search of the Receptor Ligands by the CyCLOPS (Cytometry Cell-Labeling Operable Phage Screening) Technique. Int. J. Mol. Sci. 2020, 21, 6258.
- Álvaro-Benito, M.; Freund, C. Revisiting Nonclassical HLA II Functions in Antigen Presentation: Peptide Editing and Its Modulation. HLA 2020, 96, 415–429.
- Álvaro-Benito, M.; Morrison, E.; Abualrous, E.T.; Kuropka, B.; Freund, C. Quantification of HLA-DM-Dependent Major Histocompatibility Complex of Class II Immunopeptidomes by the Peptide Landscape Antigenic Epitope Alignment Utility. Front. Immunol. 2018, 9, 872.
- Vogt, A.B.; Kropshofer, H.; Moldenhauer, G.; Hämmerling, G.J. Kinetic Analysis of Peptide Loading onto HLA-DR Molecules Mediated by HLA-DM. Proc. Natl. Acad. Sci. USA 1996, 93, 9724–9729.
- Álvaro-Benito, M.; Morrison, E.; Ebner, F.; Abualrous, E.T.; Urbicht, M.; Wieczorek, M.; Freund, C. Distinct Editing Functions of Natural HLA-DM Allotypes Impact Antigen Presentation and CD4+ T Cell Activation. Cell. Mol. Immunol. 2020, 17, 133–142.
- Reyes-Vargas, E.; Barker, A.P.; Zhou, Z.; He, X.; Jensen, P.E. HLA-DM Catalytically Enhances Peptide Dissociation by Sensing Peptide–MHC Class II Interactions throughout the Peptide-Binding Cleft. J. Biol. Chem. 2020, 295, 2959–2973.
- Poluektov, Y.; Kim, A.; Sadegh-Nasseri, S. HLA-DO and Its Role in MHC Class II Antigen Presentation. Front. Immunol. 2013, 4, 260.
- Nanaware, P.P.; Jurewicz, M.M.; Leszyk, J.D.; Shaffer, S.A.; Stern, L.J. HLA-DO Modulates the Diversity of the MHC-II Self-Peptidome. Mol. Cell. Proteom. 2019, 18, 490–503.
- Surh, C.D.; Lee, D.-S.; Fung-Leung, W.; Karlsson, L.; Sprent, J. Thymic Selection by a Single MHC/Peptide Ligand Produces a Semidiverse Repertoire of CD4+ T Cells. Immunity 1997, 7, 209–219.
- Olsson, N.; Jiang, W.; Adler, L.N.; Mellins, E.D.; Elias, J.E. Tuning DO: DM Ratios Modulates MHC Class II Immunopeptidomes. Mol. Cell. Proteom. 2022, 21, 100204.
- Zhou, Z.; Reyes-Vargas, E.; Escobar, H.; Chang, K.Y.; Barker, A.P.; Rockwood, A.L.; Delgado, J.C.; He, X.; Jensen, P.E. Peptidomic Analysis of Type 1 Diabetes Associated HLA-DQ Molecules and the Impact of HLA-DM on Peptide Repertoire Editing. Eur. J. Immunol. 2017, 47, 314–326.
- Collado, J.A.; Alvarez, I.; Ciudad, M.T.; Espinosa, G.; Canals, F.; Pujol-Borrell, R.; Carrascal, M.; Abian, J.; Jaraquemada, D. Composition of the HLA-DR-Associated Human Thymus Peptidome. Eur. J. Immunol. 2013, 43, 2273–2282.
- Nielsen, M.; Justesen, S.; Lund, O.; Lundegaard, C.; Buus, S. NetMHCIIpan-2.0—Improved Pan-Specific HLA-DR Predictions Using a Novel Concurrent Alignment and Weight Optimization Training Procedure. Immunome Res. 2010, 6, 9.
- Muixí, L.; Carrascal, M.; Alvarez, I.; Daura, X.; Martí, M.; Armengol, M.P.; Pinilla, C.; Abian, J.; Pujol-Borrell, R.; Jaraquemada, D. Thyroglobulin Peptides Associate In Vivo to HLA-DR in Autoimmune Thyroid Glands. J. Immunol. 2008, 181, 795.
- Fissolo, N.; Haag, S.; de Graaf, K.L.; Drews, O.; Stevanovic, S.; Rammensee, H.G.; Weissert, R. Naturally Presented Peptides on Major Histocompatibility Complex I and II Molecules Eluted from Central Nervous System of Multiple Sclerosis Patients. Mol. Cell. Proteom. 2009, 8, 2090–2101.
- Martinsen, V.; Kursula, P. Multiple Sclerosis and Myelin Basic Protein: Insights into Protein Disorder and Disease. Amino. Acids 2022, 54, 99–109.
- Belogurov, A.A.; Kurkova, I.N.; Friboulet, A.; Thomas, D.; Misikov, V.K.; Zakharova, M.Y.; Suchkov, S.V.; Kotov, S.V.; Alehin, A.I.; Avalle, B.; et al. Recognition and Degradation of Myelin Basic Protein Peptides by Serum Autoantibodies: Novel Biomarker for Multiple Sclerosis. J. Immunol. 2008, 180, 1258–1267.
- Belogurov, A.; Zakharov, K.; Lomakin, Y.; Surkov, K.; Avtushenko, S.; Kruglyakov, P.; Smirnov, I.; Makshakov, G.; Lockshin, C.; Gregoriadis, G.; et al. CD206-Targeted Liposomal Myelin Basic Protein Peptides in Patients with Multiple Sclerosis Resistant to First-Line Disease-Modifying Therapies: A First-in-Human, Proof-of-Concept Dose-Escalation Study. Neurotherapeutics 2016, 13, 895–904.
- Nokoff, N.J.; Rewers, M.; Cree Green, M. The Interplay of Autoimmunity and Insulin Resistance in Type 1 Diabetes. Discov. Med. 2012, 13, 115–122.
- Ito, Y.; Ashenberg, O.; Pyrdol, J.; Luoma, A.M.; Rozenblatt-Rosen, O.; Hofree, M.; Christian, E.; Ferrari de Andrade, L.; Tay, R.E.; Teyton, L.; et al. Rapid CLIP Dissociation from MHC II Promotes an Unusual Antigen Presentation Pathway in Autoimmunity. J. Exp. Med. 2018, 215, 2617–2635.
- Busch, R.; Kollnberger, S.; Mellins, E.D. HLA Associations in Inflammatory Arthritis: Emerging Mechanisms and Clinical Implications. Nat. Rev. Rheumatol. 2019, 15, 364–381.
- Markovic-Plese, S.; Fukaura, H.; Zhang, J.; al-Sabbagh, A.; Southwood, S.; Sette, A.; Kuchroo, V.K.; Hafler, D.A. T Cell Recognition of Immunodominant and Cryptic Proteolipid Protein Epitopes in Humans. J. Immunol. 1995, 155, 982.
- Loyal, L.; Braun, J.; Henze, L.; Kruse, B.; Dingeldey, M.; Reimer, U.; Kern, F.; Schwarz, T.; Mangold, M.; Unger, C.; et al. Cross-Reactive CD4+ T Cells Enhance SARS-CoV-2 Immune Responses upon Infection and Vaccination. Science (1979) 2022, 374, eabh1823.
- Gao, Y.; Cai, C.; Grifoni, A.; Müller, T.R.; Niessl, J.; Olofsson, A.; Humbert, M.; Hansson, L.; Österborg, A.; Bergman, P.; et al. Ancestral SARS-CoV-2-Specific T Cells Cross-Recognize the Omicron Variant. Nat. Med. 2022, 28, 472–476.
- Molodtsov, I.A.; Kegeles, E.; Mitin, A.N.; Mityaeva, O.; Musatova, O.E.; Panova, A.E.; Pashenkov, M.V.; Peshkova, I.O.; Alsalloum, A.; Asaad, W.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)–Specific T Cells and Antibodies in Coronavirus Disease 2019 (COVID-19) Protection: A Prospective Study. Clin. Infect. Dis. 2022, 75, e1–e9.
- Yiyuan, Y.; Xiang, W.X.; Mariuzza, R.A.A. Crystal Structure of a Complete Ternary Complex of T-Cell Receptor, Peptide–MHC, and CD4. Proc. Natl. Acad. Sci. USA 2012, 109, 5405–5410.
- Zareie, P.; Farenc, C.; la Gruta, N.L. MHC Restriction: Where Are We Now? Viral. Immunol. 2020, 33, 179–187.
- Mørch, A.M.; Bálint, Š.; Santos, A.M.; Davis, S.J.; Dustin, M.L. Coreceptors and TCR Signaling—The Strong and the Weak of It. Front Cell. Dev. Biol. 2020, 8, 597627.
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466.
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T Cell Antigen Receptor Recognition of Antigen-Presenting Molecules. Annu. Rev. Immunol. 2015, 33, 169–200.
- Hennecke, J.; Carfi, A.; Wiley, D.C. Structure of a Covalently Stabilized Complex of a Human Aβ T-Cell Receptor, Influenza HA Peptide and MHC Class II Molecule, HLA-DR1. EMBO J. 2000, 19, 5611–5624.
- Hahn, M.; Nicholson, M.J.; Pyrdol, J.; Wucherpfennig, K.W. Unconventional Topology of Self Peptide–Major Histocompatibility Complex Binding by a Human Autoimmune T Cell Receptor. Nat. Immunol. 2005, 6, 490–496.
- Kato, Z.; Stern, J.N.H.; Nakamura, H.K.; Kuwata, K.; Kondo, N.; Strominger, J.L. Positioning of Autoimmune TCR-Ob.2F3 and TCR-Ob.3D1 on the MBP85–99/HLA-DR2 Complex. Proc. Natl. Acad. Sci. USA 2008, 105, 15523–15528.
- Li, Y.; Huang, Y.; Lue, J.; Quandt, J.A.; Martin, R.; Mariuzza, R.A. Structure of a Human Autoimmune TCR Bound to a Myelin Basic Protein Self-Peptide and a Multiple Sclerosis-Associated MHC Class II Molecule. EMBO J. 2005, 24, 2968–2979.
- Kong, Y.; Jing, Y.; Allard, D.; Scavuzzo, M.A.; Sprouse, M.L.; Borowiak, M.; Bettini, M.L.; Bettini, M. A Dormant T-Cell Population with Autoimmune Potential Exhibits Low Self-Reactivity and Infiltrates Islets in Type 1 Diabetes. Eur. J. Immunol. 2022, 52, 1158–1170.
- Sethi, D.K.; Schubert, D.A.; Anders, A.-K.; Heroux, A.; Bonsor, D.A.; Thomas, C.P.; Sundberg, E.J.; Pyrdol, J.; Wucherpfennig, K.W. A Highly Tilted Binding Mode by a Self-Reactive T Cell Receptor Results in Altered Engagement of Peptide and MHC. J. Exp. Med. 2011, 208, 91–102.
- Adams, J.J.; Narayanan, S.; Liu, B.; Birnbaum, M.E.; Kruse, A.C.; Bowerman, N.A.; Chen, W.; Levin, A.M.; Connolly, J.M.; Zhu, C.; et al. T Cell Receptor Signaling Is Limited by Docking Geometry to Peptide-Major Histocompatibility Complex. Immunity 2011, 35, 681–693.
- Yin, Y.; Li, Y.; Kerzic, M.C.; Martin, R.; Mariuzza, R.A. Structure of a TCR with High Affinity for Self-Antigen Reveals Basis for Escape from Negative Selection. EMBO J. 2011, 30, 1137–1148.
- Raposo, B.; Merky, P.; Lundqvist, C.; Yamada, H.; Urbonaviciute, V.; Niaudet, C.; Viljanen, J.; Kihlberg, J.; Kyewski, B.; Ekwall, O.; et al. T Cells Specific for Post-Translational Modifications Escape Intrathymic Tolerance Induction. Nat. Commun. 2018, 9, 353.
- Carayanniotis, G. Recognition of Thyroglobulin by T Cells: The Role of Iodine. Thyroid 2007, 17, 963–973.
- Kwon, E.-J.; Ju, J.H. Impact of Posttranslational Modification in Pathogenesis of Rheumatoid Arthritis: Focusing on Citrullination, Carbamylation, and Acetylation. Int. J. Mol. Sci. 2021, 22, 10576.
- Hill, J.A.; Southwood, S.; Sette, A.; Jevnikar, A.M.; Bell, D.A.; Cairns, E. Cutting Edge: The Conversion of Arginine to Citrulline Allows for a High-Affinity Peptide Interaction with the Rheumatoid Arthritis-Associated HLA-DRB1*0401 MHC Class II Molecule. J. Immunol. 2003, 171, 538.
- Scherer, H.U.; Häupl, T.; Burmester, G.R. The Etiology of Rheumatoid Arthritis. J. Autoimmun. 2020, 110, 102400.
- Lim, J.J.; Jones, C.M.; Loh, T.J.; Ting, Y.T.; Zareie, P.; Loh, K.L.; Rossjohn, J.; Felix, N.; Supi, A.; Baker, D.G. The Shared Susceptibility Epitope of HLA-DR4 Binds Citrullinated Self-Antigens and the TCR. Sci. Immunol. 2021, 6, eabe0896.
- Song, J.; Schwenzer, A.; Wong, A.; Turcinov, S.; Rims, C.; Martinez, L.R.; Arribas-Layton, D.; Gerstner, C.; Muir, V.S.; Midwood, K.S. Shared Recognition of Citrullinated Tenascin-C Peptides by T and B Cells in Rheumatoid Arthritis. JCI Insight 2021, 6, e145217.
- Ge, C.; Weisse, S.; Xu, B.; Dobritzsch, D.; Viljanen, J.; Kihlberg, J.; Do, N.-N.; Schneider, N.; Lanig, H.; Holmdahl, R.; et al. Key Interactions in the Trimolecular Complex Consisting of the Rheumatoid Arthritis—Associated DRB1 * 04:01 Molecule, the Major Glycosylated Collagen II Peptide and the T-Cell Receptor. Ann. Rheum. Dis. 2022, 81, 480.
- McGinty, J.W.; Chow, I.-T.; Greenbaum, C.; Odegard, J.; Kwok, W.W.; James, E.A. Recognition of Posttranslationally Modified GAD65 Epitopes in Subjects With Type 1 Diabetes. Diabetes 2014, 63, 3033–3040.
- Yang, M.-L.; Horstman, S.; Gee, R.; Guyer, P.; Lam, T.T.; Kanyo, J.; Perdigoto, A.L.; Speake, C.; Greenbaum, C.J.; Callebaut, A.; et al. Citrullination of Glucokinase Is Linked to Autoimmune Diabetes. Nat. Commun. 2022, 13, 1870.
- Mannering, S.I.; Harrison, L.C.; Williamson, N.A.; Morris, J.S.; Thearle, D.J.; Jensen, K.P.; Kay, T.W.H.; Rossjohn, J.; Falk, B.A.; Nepom, G.T.; et al. The Insulin A-Chain Epitope Recognized by Human T Cells Is Posttranslationally Modified. J. Exp. Med. 2005, 202, 1191–1197.
- Cao, L.; Sun, D.; Whitaker, J.N. Citrullinated Myelin Basic Protein Induces Experimental Autoimmune Encephalomyelitis in Lewis Rats through a Diverse T Cell Repertoire. J. Neuroimmunol. 1998, 88, 21–29.
- Carrillo-Vico, A.; Leech, M.D.; Anderton, S.M. Contribution of Myelin Autoantigen Citrullination to T Cell Autoaggression in the Central Nervous System. J. Immunol. 2010, 184, 2839.
- Valdivia, A.O.; Agarwal, P.K.; Bhattacharya, S.K. Myelin Basic Protein Phospholipid Complexation Likely Competes with Deimination in Experimental Autoimmune Encephalomyelitis Mouse Model. ACS Omega 2020, 5, 15454–15467.
- Maynard, J.; Petersson, K.; Wilson, D.H.; Adams, E.J.; Blondelle, S.E.; Boulanger, M.J.; Wilson, D.B.; Garcia, K.C. Structure of an Autoimmune T Cell Receptor Complexed with Class II Peptide-MHC: Insights into MHC Bias and Antigen Specificity. Immunity 2005, 22, 81–92.
- Feng, D.; Bond, C.J.; Ely, L.K.; Maynard, J.; Garcia, K.C. Structural Evidence for a Germline-Encoded T Cell Receptor–Major Histocompatibility Complex Interaction “Codon”. Nat. Immunol. 2007, 8, 975–983.
- Zamvil, S.S.; Mitchell, D.J.; Moore, A.C.; Kitamura, K.; Steinman, L.; Rothbard, J.B. T-Cell Epitope of the Autoantigen Myelin Basic Protein That Induces Encephalomyelitis. Nature 1986, 324, 258–260.
- Christen, U.; von Herrath, M.G. Infections and Autoimmunity—Good or Bad? J. Immunol. 2005, 174, 7481.
- Markovic-Plese, S.; Hemmer, B.; Zhao, Y.; Simon, R.; Pinilla, C.; Martin, R. High Level of Cross-Reactivity in Influenza Virus Hemagglutinin-Specific CD4+ T-Cell Response: Implications for the Initiation of Autoimmune Response in Multiple Sclerosis. J. Neuroimmunol. 2005, 169, 31–38.
- Hiemstra, H.S.; Schloot, N.C.; van Veelen, P.A.; Willemen, S.J.M.; Franken, K.L.M.C.; van Rood, J.J.; de Vries, R.R.P.; Chaudhuri, A.; Behan, P.O.; Drijfhout, J.W.; et al. Cytomegalovirus in Autoimmunity: T Cell Crossreactivity to Viral Antigen and Autoantigen Glutamic Acid Decarboxylase. Proc. Natl. Acad. Sci. USA 2001, 98, 3988–3991.
- Bellucci, G.; Rinaldi, V.; Buscarinu, M.C.; Reniè, R.; Bigi, R.; Pellicciari, G.; Morena, E.; Romano, C.; Marrone, A.; Mechelli, R.; et al. Multiple Sclerosis and SARS-CoV-2: Has the Interplay Started? Front. Immunol. 2021, 12, 755333.
- Satheesh, N.J.; Salloum-Asfar, S.; Abdulla, S.A. The Potential Role of COVID-19 in the Pathogenesis of Multiple Sclerosis—A Preliminary Report. Viruses 2021, 13, 2091.
- Brennan, F.M.; Smith, N.M.G.; Owen, S.; Li, C.; Amjadi, P.; Green, P.; Andersson, A.; Palfreeman, A.C.; Hillyer, P.; Foey, A.; et al. Resting CD4+ effector Memory T Cells Are Precursors of Bystander-Activated Effectors: A Surrogate Model of Rheumatoid Arthritis Synovial T-Cell Function. Arthritis Res. 2008, 10, R36.
- Martino, G.; Grohovaz, F.; Brambilla, E.; Codazzi, F.; Consiglio, A.; Clementi, E.; Filippi, M.; Comi, G.; Grimaldi, L.M.E. Proinflammatory Cytokines Regulate Antigen-Independent T-Cell Activation by Two Separate Calcium-Signaling Pathways in Multiple Sclerosis Patients. Ann. Neurol. 1998, 43, 340–349.
- Steimle, V.; Siegrist, C.-A.; Mottet, A.; Lisowska-Grospierre, B.; Mach, B. Regulation of MHC Class II Expression by Interferon-γ Mediated by the Transactivator Gene CIITA. Science (1979) 1994, 265, 106–109.
- Iglesias, B.M.; Cerase, J.; Ceracchini, C.; Levi, G.; Aloisi, F. Analysis of B7-1 and B7-2 Costimulatory Ligands in Cultured Mouse Microglia: Upregulation by Interferon-γ and Lipopolysaccharide and Downregulation by Interleukin-10, Prostaglandin E2 and Cyclic AMP-Elevating Agents. J. Neuroimmunol. 1997, 72, 83–93.
- Nikcevich, K.M.; Gordon, K.B.; Tan, L.; Hurst, S.D.; Kroepfl, J.F.; Gardinier, M.; Barrett, T.A.; Miller, S.D. IFN-Gamma-Activated Primary Murine Astrocytes Express B7 Costimulatory Molecules and Prime Naive Antigen-Specific T Cells. J. Immunol. 1997, 158, 614.
- Russell, M.A.; Redick, S.D.; Blodgett, D.M.; Richardson, S.J.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; Bottino, R.; Brissova, M.; Spaeth, J.M.; et al. HLA Class II Antigen Processing and Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of Islet β-Cells From Donors With Type 1 Diabetes. Diabetes 2019, 68, 988–1001.
- Quesada-Masachs, E.; Zilberman, S.; Rajendran, S.; Chu, T.; McArdle, S.; Kiosses, W.B.; Lee, J.-H.M.; Yesildag, B.; Benkahla, M.A.; Pawlowska, A.; et al. Upregulation of HLA Class II in Pancreatic Beta Cells from Organ Donors with Type 1 Diabetes. Diabetologia 2022, 65, 387–401.
- Zhao, Y.; Scott, N.A.; Quah, H.S.; Krishnamurthy, B.; Bond, F.; Loudovaris, T.; Mannering, S.I.; Kay, T.W.H.; Thomas, H.E. Mouse Pancreatic Beta Cells Express MHC Class II and Stimulate CD4+ T Cells to Proliferate. Eur. J. Immunol. 2015, 45, 2494–2503.
- Scott, N.A.; Zhao, Y.; Krishnamurthy, B.; Mannering, S.I.; Kay, T.W.H.; Thomas, H.E. IFNγ-Induced MHC Class II Expression on Islet Endothelial Cells Is an Early Marker of Insulitis but Is Not Required for Diabetogenic CD4+ T Cell Migration. Front. Immunol. 2018, 9, 2800.
- Gobin, S.J.P.; Montagne, L.; van Zutphen, M.; van der Valk, P.; van den Elsen, P.J.; de Groot, C.J.A. Upregulation of Transcription Factors Controlling MHC Expression in Multiple Sclerosis Lesions. Glia 2001, 36, 68–77.
- Falcão, A.M.; van Bruggen, D.; Marques, S.; Meijer, M.; Jäkel, S.; Agirre, E.; Samudyata; Floriddia, E.M.; Vanichkina, D.P.; ffrench-Constant, C.; et al. Disease-Specific Oligodendrocyte Lineage Cells Arise in Multiple Sclerosis. Nat. Med. 2018, 24, 1837–1844.
- Kanazawa, S.; Ota, S.; Sekine, C.; Tada, T.; Otsuka, T.; Okamoto, T.; Sønderstrup, G.; Peterlin, B.M. Aberrant MHC Class II Expression in Mouse Joints Leads to Arthritis with Extraarticular Manifestations Similar to Rheumatoid Arthritis. Proc. Natl. Acad. Sci. USA 2006, 103, 14465–14470.
- Fasano, R.; Malerba, E.; Prete, M.; Solimando, A.G.; Buonavoglia, A.; Silvestris, N.; Leone, P.; Racanelli, V. Impact of Antigen Presentation Mechanisms on Immune Response in Autoimmune Hepatitis. Front. Immunol. 2022, 12, 814155.
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.-S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.-S.; Bartley, C.M.; et al. Clonally Expanded B Cells in Multiple Sclerosis Bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327.
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms. Viruses 2019, 11, 762.
- Tengvall, K.; Huang, J.; Hellström, C.; Kammer, P.; Biström, M.; Ayoglu, B.; Bomfim, I.L.; Stridh, P.; Butt, J.; Brenner, N.; et al. Molecular Mimicry between Anoctamin 2 and Epstein-Barr Virus Nuclear Antigen 1 Associates with Multiple Sclerosis Risk. Proc. Natl. Acad. Sci. USA 2019, 116, 16955–16960.
- Wucherpfennig, K.W.; Strominger, J.L. Molecular Mimicry in T Cell-Mediated Autoimmunity: Viral Peptides Activate Human T Cell Clones Specific for Myelin Basic Protein. Cell 1995, 80, 695–705.
- Lünemann, J.D.; Jelčić, I.; Roberts, S.; Lutterotti, A.; Tackenberg, B.; Martin, R.; Munz, C. EBNA1-Specific T Cells from Patients with Multiple Sclerosis Cross React with Myelin Antigens and Co-Produce IFN-γ and IL-2. J. Exp. Med. 2008, 205, 1763–1773.
- Zdimerova, H.; Murer, A.; Engelmann, C.; Raykova, A.; Deng, Y.; Gujer, C.; Rühl, J.; McHugh, D.; Caduff, N.; Naghavian, R.; et al. Attenuated Immune Control of Epstein–Barr Virus in Humanized Mice Is Associated with the Multiple Sclerosis Risk Factor HLA-DR15. Eur. J. Immunol. 2021, 51, 64–75.
- Latorre, D.; Kallweit, U.; Armentani, E.; Foglierini, M.; Mele, F.; Cassotta, A.; Jovic, S.; Jarrossay, D.; Mathis, J.; Zellini, F.; et al. T Cells in Patients with Narcolepsy Target Self-Antigens of Hypocretin Neurons. Nature 2018, 562, 63–68.
- Luo, G.; Ambati, A.; Lin, L.; Bonvalet, M.; Partinen, M.; Ji, X.; Maecker, H.T.; Mignot, E.J.-M. Autoimmunity to Hypocretin and Molecular Mimicry to Flu in Type 1 Narcolepsy. Proc. Natl. Acad. Sci. USA 2018, 115, E12323–E12332.
- Vuorela, A.; Freitag, T.L.; Leskinen, K.; Pessa, H.; Härkönen, T.; Stracenski, I.; Kirjavainen, T.; Olsen, P.; Saarenpää-Heikkilä, O.; Ilonen, J.; et al. Enhanced Influenza A H1N1 T Cell Epitope Recognition and Cross-Reactivity to Protein-O-Mannosyltransferase 1 in Pandemrix-Associated Narcolepsy Type 1. Nat. Commun. 2021, 12, 2283.
- Koning, D.; Quakkelaar, E.D.; Schellens, I.M.M.; Spierings, E.; van Baarle, D. Protective HLA Alleles Recruit Biased and Largely Similar Antigen-Specific T Cell Repertoires across Different Outcomes in HIV Infection. J. Immunol. 2022, 208, 3–15.
- Abualrous, E.T.; Sticht, J.; Freund, C. Major Histocompatibility Complex (MHC) Class I and Class II Proteins: Impact of Polymorphism on Antigen Presentation. Curr. Opin. Immunol. 2021, 70, 95–104.
- Poomarimuthu, M.; Ramasamy, T.; Govindan, R.; Andiappan, R.; Nagarajan, G.; Kadiam, S.; Mariakuttikan, J. Association of HLA-DRB1 Alleles with Rheumatic Fever and Rheumatic Heart Disease: A Meta-Analysis. Immunol. Investig. 2022, 51, 221–232.
- Panhuber, A.; Lamorte, G.; Bruno, V.; Cetin, H.; Bauer, W.; Höftberger, R.; Erber, A.C.; Frommlet, F.; Koneczny, I. A Systematic Review and Meta-Analysis of HLA Class II Associations in Patients with IgG4 Autoimmunity. Sci. Rep. 2022, 12, 9229.
- Zhao, L.P.; Papadopoulos, G.K.; Moustakas, A.K.; Bondinas, G.P.; Carlsson, A.; Larsson, H.E.; Ludvigsson, J.; Marcus, C.; Persson, M.; Samuelsson, U.; et al. Nine Residues in HLA-DQ Molecules Determine with Susceptibility and Resistance to Type 1 Diabetes among Young Children in Sweden. Sci. Rep. 2021, 11, 8821.
- Zhao, L.P.; Papadopoulos, G.K.; Kwok, W.W.; Moustakas, A.K.; Bondinas, G.P.; Carlsson, A.; Elding Larsson, H.; Ludvigsson, J.; Marcus, C.; Samuelsson, U.; et al. Next-Generation HLA Sequence Analysis Uncovers Seven HLA-DQ Amino Acid Residues and Six Motifs Resistant to Childhood Type 1 Diabetes. Diabetes 2020, 69, 2523–2535.
- Jones, E.Y.; Fugger, L.; Strominger, J.L.; Siebold, C. MHC Class II Proteins and Disease: A Structural Perspective. Nat. Rev. Immunol. 2006, 6, 271–282.
More
Information
Subjects:
Immunology; Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
5.3K
Revisions:
3 times
(View History)
Update Date:
02 Feb 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No