 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gaetano Pacinella | -- | 4703 | 2023-01-30 11:44:31 | | | |
| 2 | Rita Xu | Meta information modification | 4703 | 2023-01-31 06:21:40 | | |
Video Upload Options
Vascular diseases of the elderly are a topic of enormous interest in clinical practice, as they have great epidemiological significance and lead to ever-increasing healthcare expenditures. The mechanisms underlying these pathologies have been increasingly characterized. It has emerged that endothelial dysfunction and chronic inflammation play a diriment role among the most relevant pathophysiological mechanisms. As one can easily imagine, various processes occur during aging, and several pathways undergo irreversible alterations that can promote the decline and aberrations that trigger the diseases above. Endothelial dysfunction and aging of circulating and resident cells are the main characteristics of the aged organism; they represent the framework within which an enormous array of molecular abnormalities occur and contribute to accelerating and perpetuating the decline of organs and tissues.
1. Introduction
2. Endothelial Dysfunction and Impaired Angiogenesis in the Elderly
2.1. Endothelial Dysfunction in Hypertension
Causative Mechanisms of Endothelial Alterations in Hypertension
2.2. Endothelial Dysfunction in Diabetes
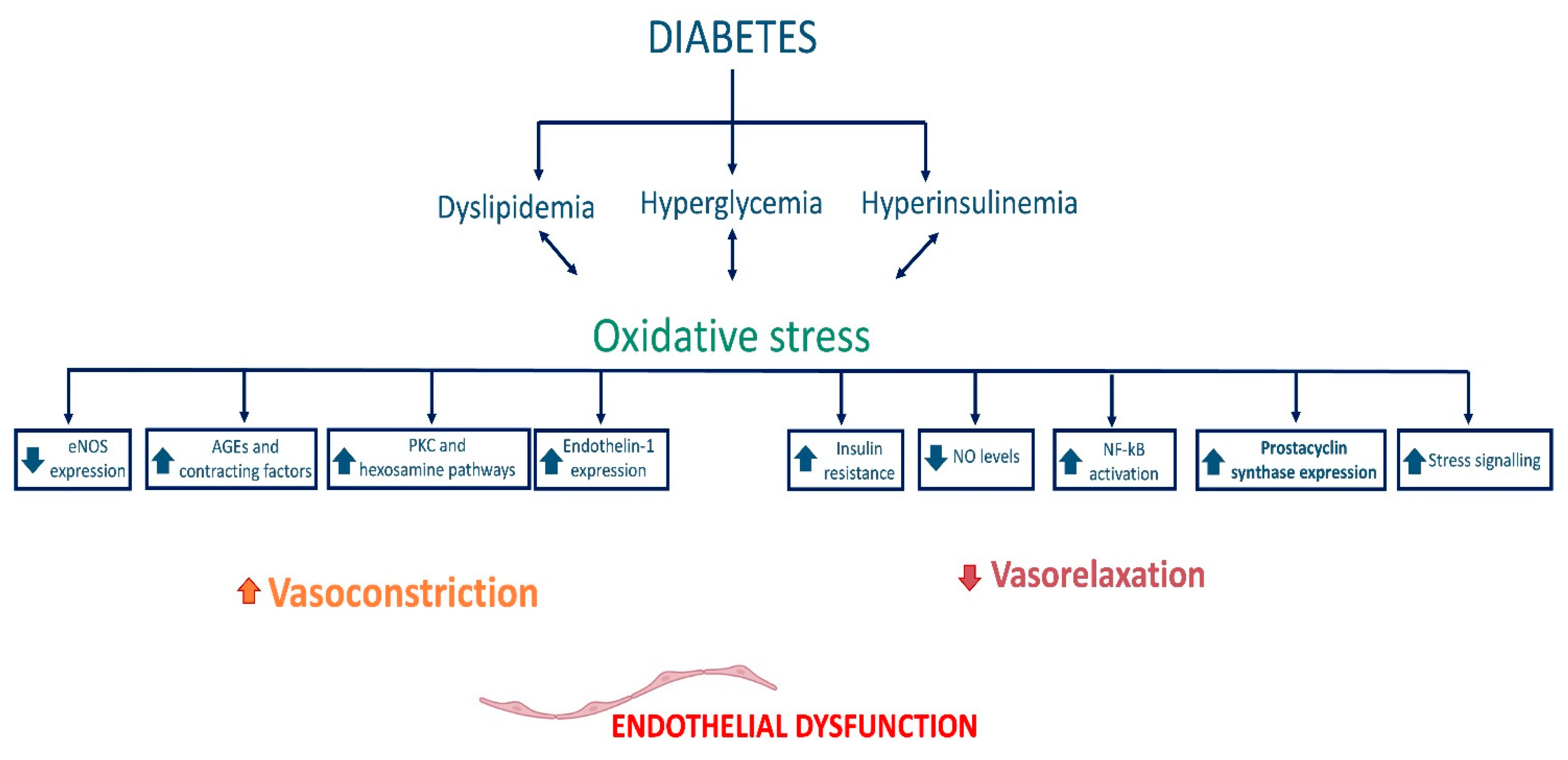
2.3. Endothelial Dysfunction in Cerebrovascular Diseases
References
- Vatner, S.F.; Zhang, J.; Vyzas, C.; Mishra, K.; Graham, R.M.; Vatner, D.E. Vascular Stiffness in Aging and Disease. Front. Physiol. 2021, 12, 762437.
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ. Res. 2018, 123, 825–848.
- Filippini, A.; Tamagnone, L.; D’Alessio, A. Endothelial Cell Metabolism in Vascular Functions. Cancers 2022, 14, 1929.
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial Dysfunction: A Comprehensive Appraisal. Cardiovasc. Diabetol. 2006, 5, 4.
- Herrera, M.D.; Mingorance, C.; Rodríguez-Rodríguez, R.; Alvarez de Sotomayor, M. Endothelial Dysfunction and Aging: An Update. Ageing Res. Rev. 2010, 9, 142–152.
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.D.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial Dysfunction and Angiogenesis Impairment in the Ageing Vasculature. Nat. Rev. Cardiol. 2018, 15, 555–565.
- Tesauro, M.; Mauriello, A.; Rovella, V.; Annicchiarico-Petruzzelli, M.; Cardillo, C.; Melino, G.; Di Daniele, N. Arterial Ageing: From Endothelial Dysfunction to Vascular Calcification. J. Intern. Med. 2017, 281, 471–482.
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217.
- Ataei Ataabadi, E.; Golshiri, K.; Jüttner, A.A.; de Vries, R.; Van den Berg-Garrelds, I.; Nagtzaam, N.M.A.; Khan, H.N.; Leijten, F.P.J.; Brandt, R.M.C.; Dik, W.A.; et al. Soluble Guanylate Cyclase Activator BAY 54-6544 Improves Vasomotor Function and Survival in an Accelerated Ageing Mouse Model. Aging Cell. 2022, 21, e13683.
- Golshiri, K.; Ataei Ataabadi, E.; Rubio-Beltran, E.; Dutheil, S.; Yao, W.; Snyder, G.L.; Davis, R.E.; van der Pluijm, I.; Brandt, R.; Van den Berg-Garrelds, I.M.; et al. Selective Phosphodiesterase 1 Inhibition Ameliorates Vascular Function, Reduces Inflammatory Response, and Lowers Blood Pressure in Aging Animals. J. Pharmacol. Exp. Ther. 2021, 378, 173–183.
- Brew, B.J.; McArthur, J.C. “A Man Is as Old as His Arteries” (Attributed to Thomas Sydenham, the English Hippocrates). AIDS 2020, 34, 637–639.
- Sewduth, R.; Santoro, M.M. “Decoding” Angiogenesis: New Facets Controlling Endothelial Cell Behavior. Front. Physiol. 2016, 7, 306.
- Eilken, H.M.; Adams, R.H. Dynamics of Endothelial Cell Behavior in Sprouting Angiogenesis. Curr. Opin. Cell. Biol. 2010, 22, 617–625.
- Adams, R.H.; Alitalo, K. Molecular Regulation of Angiogenesis and Lymphangiogenesis. Nat. Rev. Mol. Cell. Biol. 2007, 8, 464–478.
- Ramasamy, S.K.; Kusumbe, A.P.; Wang, L.; Adams, R.H. Endothelial Notch Activity Promotes Angiogenesis and Osteogenesis in Bone. Nature 2014, 507, 376–380.
- Mühleder, S.; Fernández-Chacón, M.; Garcia-Gonzalez, I.; Benedito, R. Endothelial Sprouting, Proliferation, or Senescence: Tipping the Balance from Physiology to Pathology. Cell. Mol. Life Sci. 2021, 78, 1329–1354.
- Gerhardt, H.; Betsholtz, C. Endothelial-Pericyte Interactions in Angiogenesis. Cell. Tissue Res. 2003, 314, 15–23.
- Rivard, A.; Fabre, J.E.; Silver, M.; Chen, D.; Murohara, T.; Kearney, M.; Magner, M.; Asahara, T.; Isner, J.M. Age-Dependent Impairment of Angiogenesis. Circulation 1999, 99, 111–120.
- Chigogidze, M.; Sharashidze, N.; Pagava, Z.; Taboridze, I. The Correlation of Collateral Circulation and Age during Acute Ischemic Heart Disease. Eur. Sci. J. ESJ 2020, 16, 335.
- Sadoun, E.; Reed, M.J. Impaired Angiogenesis in Aging Is Associated with Alterations in Vessel Density, Matrix Composition, Inflammatory Response, and Growth Factor Expression. J. Histochem. Cytochem. 2003, 51, 1119–1130.
- Theories and Molecular Basis of Vascular Aging: A Review of the Literature from VascAgeNet Group on Pathophysiological Mechanisms of Vascular Aging—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/35955804/ (accessed on 9 November 2022).
- Iakovou, E.; Kourti, M. A Comprehensive Overview of the Complex Role of Oxidative Stress in Aging, The Contributing Environmental Stressors and Emerging Antioxidant Therapeutic Interventions. Front. Aging Neurosci. 2022, 14, 827900.
- Leyane, T.S.; Jere, S.W.; Houreld, N.N. Oxidative Stress in Ageing and Chronic Degenerative Pathologies: Molecular Mechanisms Involved in Counteracting Oxidative Stress and Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 7273.
- Wagatsuma, A. Effect of Aging on Expression of Angiogenesis-Related Factors in Mouse Skeletal Muscle. Exp. Gerontol. 2006, 41, 49–54.
- Wang, H.; Keiser, J.A.; Olszewski, B.; Rosebury, W.; Robertson, A.; Kovesdi, I.; Gordon, D. Delayed Angiogenesis in Aging Rats and Therapeutic Effect of Adenoviral Gene Transfer of VEGF. Int. J. Mol. Med. 2004, 13, 581–587.
- Peters, A.; Josephson, K.; Vincent, S.L. Effects of Aging on the Neuroglial Cells and Pericytes within Area 17 of the Rhesus Monkey Cerebral Cortex. Anat. Rec. 1991, 229, 384–398.
- Burns, E.M.; Kruckeberg, T.W.; Gaetano, P.K. Changes with Age in Cerebral Capillary Morphology. Neurobiol. Aging 1981, 2, 283–291.
- Yamamoto, N.; Oyaizu, T.; Enomoto, M.; Horie, M.; Yuasa, M.; Okawa, A.; Yagishita, K. VEGF and BFGF Induction by Nitric Oxide Is Associated with Hyperbaric Oxygen-Induced Angiogenesis and Muscle Regeneration. Sci. Rep. 2020, 10, 2744.
- Jüttner, A.A.; Danser, A.H.J.; Roks, A.J.M. Pharmacological Developments in Antihypertensive Treatment through Nitric Oxide-CGMP Modulation. Adv. Pharmacol. 2022, 94, 57–94.
- Chin, L.C.; Kumar, P.; Palmer, J.A.; Rophael, J.A.; Dolderer, J.H.; Thomas, G.P.L.; Morrison, W.A.; Penington, A.J.; Stewart, A.G.; Mitchell, G.M. The Influence of Nitric Oxide Synthase 2 on Cutaneous Wound Angiogenesis. Br. J. Dermatol. 2011, 165, 1223–1235.
- Bachetti, T.; Comini, L.; Curello, S.; Bastianon, D.; Palmieri, M.; Bresciani, G.; Callea, F.; Ferrari, R. Co-Expression and Modulation of Neuronal and Endothelial Nitric Oxide Synthase in Human Endothelial Cells. J. Mol. Cell. Cardiol. 2004, 37, 939–945.
- Rocha, B.S. The Nitrate-Nitrite-Nitric Oxide Pathway on Healthy Ageing: A Review of Pre-Clinical and Clinical Data on the Impact of Dietary Nitrate in the Elderly. Front. Aging 2021, 2, 778467.
- Hodges, N.A.; Suarez-Martinez, A.D.; Murfee, W.L. Understanding Angiogenesis during Aging: Opportunities for Discoveries and New Models. J. Appl. Physiol. (1985) 2018, 125, 1843–1850.
- Oliveros, E.; Patel, H.; Kyung, S.; Fugar, S.; Goldberg, A.; Madan, N.; Williams, K.A. Hypertension in Older Adults: Assessment, Management, and Challenges. Clin. Cardiol. 2020, 43, 99–107.
- Lloyd-Jones, D.M.; Evans, J.C.; Levy, D. Hypertension in Adults across the Age Spectrum: Current Outcomes and Control in the Community. JAMA 2005, 294, 466–472.
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial Fluid Shear Stress Sensing in Vascular Health and Disease. J. Clin. Investig. 2016, 126, 821–828.
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial Responses to Shear Stress in Atherosclerosis: A Novel Role for Developmental Genes. Nat. Rev. Cardiol. 2020, 17, 52–63.
- Chala, N.; Moimas, S.; Giampietro, C.; Zhang, X.; Zambelli, T.; Exarchos, V.; Nazari-Shafti, T.Z.; Poulikakos, D.; Ferrari, A. Mechanical Fingerprint of Senescence in Endothelial Cells. Nano Lett. 2021, 21, 4911–4920.
- Warboys, C.M.; de Luca, A.; Amini, N.; Luong, L.; Duckles, H.; Hsiao, S.; White, A.; Biswas, S.; Khamis, R.; Chong, C.K.; et al. Disturbed Flow Promotes Endothelial Senescence via a P53-Dependent Pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 985–995.
- Zhu, S.; Dong, Z.; Ke, X.; Hou, J.; Zhao, E.; Zhang, K.; Wang, F.; Yang, L.; Xiang, Z.; Cui, H. The Roles of Sirtuins Family in Cell Metabolism during Tumor Development. Semin. Cancer Biol. 2019, 57, 59–71.
- Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/36077539/ (accessed on 9 November 2022).
- Zhou, S.; Chen, H.-Z.; Wan, Y.-Z.; Zhang, Q.-J.; Wei, Y.-S.; Huang, S.; Liu, J.-J.; Lu, Y.-B.; Zhang, Z.-Q.; Yang, R.-F.; et al. Repression of P66Shc Expression by SIRT1 Contributes to the Prevention of Hyperglycemia-Induced Endothelial Dysfunction. Circ. Res. 2011, 109, 639–648.
- Takaishi, K.; Kinoshita, H.; Kawashima, S.; Kawahito, S. Human Vascular Smooth Muscle Function and Oxidative Stress Induced by NADPH Oxidase with the Clinical Implications. Cells 2021, 10, 1947.
- Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS?—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/31382355/ (accessed on 9 November 2022).
- Wang, M.; Zhang, J.; Walker, S.J.; Dworakowski, R.; Lakatta, E.G.; Shah, A.M. Involvement of NADPH Oxidase in Age-Associated Cardiac Remodeling. J. Mol. Cell. Cardiol. 2010, 48, 765–772.
- Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/35127755/ (accessed on 9 November 2022).
- Sahoo, S.; Meijles, D.N.; Pagano, P.J. NADPH Oxidases: Key Modulators in Aging and Age-Related Cardiovascular Diseases? Clin. Sci. 2016, 130, 317–335.
- Fountain, J.H.; Lappin, S.L. Physiology, Renin Angiotensin System. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022.
- Jaimes, E.A.; Galceran, J.M.; Raij, L. Angiotensin II Induces Superoxide Anion Production by Mesangial Cells. Kidney Int. 1998, 54, 775–784.
- Yang, J.-X.; Pan, Y.-Y.; Wang, X.-X.; Qiu, Y.-G.; Mao, W. Endothelial Progenitor Cells in Age-Related Vascular Remodeling. Cell. Transplant. 2018, 27, 786–795.
- Umemura, T.; Higashi, Y. Endothelial Progenitor Cells: Therapeutic Target for Cardiovascular Diseases. J. Pharmacol. Sci. 2008, 108, 1–6.
- Benetos, A.; Petrovic, M.; Strandberg, T. Hypertension Management in Older and Frail Older Patients. Circ. Res. 2019, 124, 1045–1060.
- Eckel, R.H.; Kahn, S.E.; Ferrannini, E.; Goldfine, A.B.; Nathan, D.M.; Schwartz, M.W.; Smith, R.J.; Smith, S.R. Obesity and Type 2 Diabetes: What Can Be Unified and What Needs to Be Individualized? J. Clin. Endocrinol. Metab. 2011, 96, 1654–1663.
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell. Metab. 2019, 30, 129–142.e4.
- Sulistyowati, E.; Permatasari, N.; Aris Widodo, M. Combined Effects of Shear Stress and Glucose on the Morphology, Actin Filaments, and VE-Cadherin of Endothelial Cells in Vitro. Int. J. Cardiol. Heart Vasc. 2017, 15, 31–35.
- Saltiel, A.R.; Kahn, C.R. Insulin Signalling and the Regulation of Glucose and Lipid Metabolism. Nature 2001, 414, 799–806.
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625.
- Schmidt, A.M.; Yan, S.D.; Wautier, J.L.; Stern, D. Activation of Receptor for Advanced Glycation End Products: A Mechanism for Chronic Vascular Dysfunction in Diabetic Vasculopathy and Atherosclerosis. Circ. Res. 1999, 84, 489–497.
- Chavakis, T.; Bierhaus, A.; Nawroth, P.P. RAGE (Receptor for Advanced Glycation End Products): A Central Player in the Inflammatory Response. Microbes Infect. 2004, 6, 1219–1225.
- Chakravarthy, U.; Hayes, R.G.; Stitt, A.W.; McAuley, E.; Archer, D.B. Constitutive Nitric Oxide Synthase Expression in Retinal Vascular Endothelial Cells Is Suppressed by High Glucose and Advanced Glycation End Products. Diabetes 1998, 47, 945–952.
- Xu, B.; Chibber, R.; Ruggiero, D.; Kohner, E.; Ritter, J.; Ferro, A.; Ruggerio, D. Impairment of Vascular Endothelial Nitric Oxide Synthase Activity by Advanced Glycation End Products. FASEB J. 2003, 17, 1289–1291.
- Brownlee, M. Biochemistry and Molecular Cell Biology of Diabetic Complications. Nature 2001, 414, 813–820.
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High Glucose Level and Free Fatty Acid Stimulate Reactive Oxygen Species Production through Protein Kinase C—Dependent Activation of NAD(P)H Oxidase in Cultured Vascular Cells. Diabetes 2000, 49, 1939–1945.
- Boden, G.; She, P.; Mozzoli, M.; Cheung, P.; Gumireddy, K.; Reddy, P.; Xiang, X.; Luo, Z.; Ruderman, N. Free Fatty Acids Produce Insulin Resistance and Activate the Proinflammatory Nuclear Factor-KappaB Pathway in Rat Liver. Diabetes 2005, 54, 3458–3465.
- Buford, T.W. Hypertension and Aging. Ageing Res. Rev. 2016, 26, 96–111.
- Del Cuore, A.; Pacinella, G.; Riolo, R.; Tuttolomondo, A. The Role of Immunosenescence in Cerebral Small Vessel Disease: A Review. Int. J. Mol. Sci. 2022, 23, 7136.
- Pierce, G.L.; Lesniewski, L.A.; Lawson, B.R.; Beske, S.D.; Seals, D.R. Nuclear Factor-κB Activation Contributes to Vascular Endothelial Dysfunction via Oxidative Stress in Overweight/Obese Middle-Aged and Older Humans. Circulation 2009, 119, 1284–1292.
- Wardlaw, J.M.; Sandercock, P.a.G.; Dennis, M.S.; Starr, J. Is Breakdown of the Blood-Brain Barrier Responsible for Lacunar Stroke, Leukoaraiosis, and Dementia? Stroke 2003, 34, 806–812.
- Henning, E.C.; Warach, S.; Spatz, M. Hypertension-Induced Vascular Remodeling Contributes to Reduced Cerebral Perfusion and the Development of Spontaneous Stroke in Aged SHRSP Rats. J. Cereb. Blood Flow Metab. 2010, 30, 827–836.
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302.

