Vascular diseases of the elderly are a topic of enormous interest in clinical practice, as they have great epidemiological significance and lead to ever-increasing healthcare expenditures. The mechanisms underlying these pathologies have been increasingly characterized. It has emerged that endothelial dysfunction and chronic inflammation play a diriment role among the most relevant pathophysiological mechanisms. As one can easily imagine, various processes occur during aging, and several pathways undergo irreversible alterations that can promote the decline and aberrations that trigger the diseases above. Endothelial dysfunction and aging of circulating and resident cells are the main characteristics of the aged organism; they represent the framework within which an enormous array of molecular abnormalities occur and contribute to accelerating and perpetuating the decline of organs and tissues.
- endothelial dysfunction
- cellular senescence
- inflamm-ageing
- vascular alterations
- chronic inflammation
1. Introduction
2. Endothelial Dysfunction and Impaired Angiogenesis in the Elderly
Age-related endothelial dysfunction is associated with the structural impairment of the circulatory system. Microvascular homeostasis results from the dynamic balance between pro-angiogenic and anti-angiogenic factors. Accumulating evidence suggests that aging is characterized by altered angiogenic processes leading to a progressive deterioration of microvascular homeostasis and thus is predisposed to the development of cardiovascular diseases, such as hypertension, diabetes, atherosclerosis, and cerebrovascular diseases. Angiogenesis, defined as forming new vessels from pre-existing functional vessels, is necessary for physiological and pathological conditions. It is essential during embryogenesis, cardiovascular maturation, and tissue development and repair [13][43]. It also involves many pathological processes, such as tumor progression and chronic inflammation. Angiogenesis is a dynamic, complex mechanism involving several actors, such as endothelial cells, pericytes, and growth factors; it occurs in tiny capillaries and implicates the sprouting of existing endothelial cells. Specifically, the driving mechanism is the arrangement of endothelial cells in tip and stalk cells. Tip cells make up filopodia, which invade the surrounding tissue guiding the formation of new vessels [14][44]. This process is regulated by the interplay between the Vascular Endothelial Growth Factor (VEGF) and Notch signaling [15][45]. Tip cells abundantly express receptors for VEGF (VEGFR), and the activation of the VEGF pathway leads to filopodia formation, conferring the sprouting phenotype to tip cells. However, the VEGF pathway also induces the activation of the Notch signaling in the neighbouring stalk cells, which inhibits the VEGFR expression, conferring them the non-sprouting phenotype [16][46]. Thus, tip cells are characterized by high VEGF and low Notch expression, while the stalk cells have low VEGF and high Notch expression. Notch also has a pivotal role in lumen morphogenesis and the acquisition of barrier characteristics. Under physiological conditions, the balance between VEGF and Notch signaling is strictly regulated, forming organized structures. Beyond VEGF and Notch pathways, several factors, directly or indirectly, control endothelial proliferation, sprouting, and maturation. Among these, the most important are Ang-Tie, BMP/TGF-β, EphrinB2-EphB4, Cxcr4-Cxcl12, and Wnt [17][47]. Pericytes also represent important players in angiogenesis by modulating endothelial cell proliferation and migration [18][48]. Additionally, they have a crucial role in regulating endothelial dynamics and capillary function. Noteworthy, the angiogenic function is progressively lost during aging, as evidenced in animal trials and clinical studies. For example, experimental models showed that old animals have a reduced capability for collateral vessel development in response to ischemia [19][49]. Similarly, older patients with coronary artery disease (especially arterial obstructive vascular disease) and consequent congestive heart failure have a reduced capacity to form collateral arteries [20][50]. Additionally, in vitro studies showed that endothelial cells from old mice have reduced proliferating and migrating capacity [21][51]. Another exciting piece of evidence is that, although cancer incidence increases with age, the progression is slowed down in older individuals compared to younger individuals. This could be partly explained by the impairment of angiogenesis, which represents the driving process for tumor growth and metastasis, as confirmed in experimental studies. Several mechanisms have been hypothesized to underpin the age-related impairment of angiogenesis. First, the senescence of endothelial cells is associated with significant changes in gene expression, cellular replication, and morphological phenotypes, compromising the endothelium integrity and, consequently, the angiogenesis. The molecular mechanisms underlying endothelial cell senescence have yet to be fully understood. However, cell cycle deregulation, oxidative stress, impaired calcium signaling, and inflammation could be involved. Robust evidence highlights a prominent role, specifically in oxidative stress. It is defined as the consequence of the imbalance between the production and removal of reactive oxygen and nitrogen species (ROS and RNS, respectively) [22][52]. Much evidence has confirmed that free radicals are dangerous, and many mechanisms have been proposed to elucidate the relationship between oxidative stress and aging [23][53]. ROS and RNS include unstable free radicals, such as hydroxyl (OH−), superoxide anions (O2−), nitric oxide (NO−) radicals, and non-free radicals, such as hydrogen peroxide (H2O2). They are highly reactive, unstable species and can modify several substrates, including DNA, proteins, and lipids. The primary source of ROS is mitochondria, but peroxisomes, microsomes, and immune system cells also contribute to their production. Under physiological conditions, ROS plays an essential biological function within different cellular processes. However, the increase in their concentrations has several adverse effects when inducing impaired cell structure and functions, resulting in senescence [24][54]. The increase in ROS is generally neutralized by antioxidants, which can be classified as enzymatic, such as superoxide dismutase (SOD), glutathione peroxidase (GPX), catalase (CAT), and thioredoxin (Trx), and non-enzymatic, such as carotenoids, ubiquinol, and flavonoids. The imbalance between ROS and antioxidants leads to oxidative stress. Another important mechanism involved in age-related impaired angiogenesis is the alteration of growth factors. Experimental studies showed that old mice had reduced levels of VEGF and capillary-to-fiber perimeter exchange than young mice [25][55]. The restoration of VEGF signaling in aged animals by transfecting the VEGF121 gene could rescue the age-dependent decline in angiogenesis [26][56]. Overall, cumulating literature evidence shows that aging is characterized by the reduction of VEGF levels, which could be due to the impaired activation of the transcription factor for VEGF, namely hypoxia-inducible factor-1α [25][55]. Interestingly, pericytes could also contribute to the impairment of angiogenesis. During aging, pericytes undergo ultrastructural changes that could compromise their function [27][57]. Some authors also described a reduction in pericytes in aged monkeys and rats. In contrast, others reported increased pericytes or no changes [27][28][57,58]. Thus, findings on the possible role of pericytes in age-related endothelial and angiogenic dysfunction are still controversial, and further studies are required to elucidate them. The decrease of nitric oxide (NO) is another hallmark of aging, with a pivotal role in endothelial dysfunction and angiogenic alteration. NO is a crucial regulator of endothelial homeostasis and has multiple biological functions, including stimulating VEGF secretion from endothelial cells and macrophages [29][59]. Chin et al., in in vivo murine models, showed that the inhibition of endothelial nitric oxide synthase (eNOS) was associated with the impairment of angiogenesis not because of aging per se but because of all the mechanisms (including endothelial dysfunction, smooth muscle cell disruption, increased oxidative stress, and DNA damage) that mark it [30][31][60,61]. Although eNOS represents the primary source of NO in the vascular endothelium, neuronal NOS (nNOS) also contributes to endothelial cells-derived NO. Indeed, the constitutive expression of nNOS has also been described in vascular endothelial cells [32][33][62,63]. Similarly to eNOS, nNOS decreases during aging, contributing to the age-related reduction of NO. In summary, several mechanisms have been proposed to explain the relationship among endothelial dysfunction, impaired angiogenesis, and aging, but there is a long way to go. The discovery of mechanisms underpinnings age-related angiogenesis impairment is hampered by the difficulty in obtaining reliable in vivo and in vitro that can reflect the authentic setting of the elderly [34][64]. Indeed, there is no gold standard for angiogenic models in the field of aging research. Many questions remain to be answered, as brilliantly described in the review by Hodges et al.2.1. Endothelial Dysfunction in Hypertension
Arterial hypertension, defined as a persistent increase in arterial pressure, represents a leading modifiable risk factor for cardiovascular diseases and premature death worldwide. Furthermore, the global burden of hypertension is growing due to population aging, which is expected to affect one-third of the population worldwide by 2025 [35][65]. Hypertension is a multifactorial disease, and the pathogenesis results from the interaction have genetic, epigenetic, and environmental factors: among these, age is the most critical risk factor. Epidemiological studies revealed that the prevalence of hypertension increases from 27% in patients younger than sixty years old to 74% in those older than eighty years old [36][66]. Ground-breaking discoveries on the molecular mechanisms of ageing have unveiled several mechanisms involved in age-related hypertension: endothelial dysfunction has a key mechanistic role in initiating and maintaining arterial hypertension. The endothelial function is mainly regulated by molecular mediators, such as NO and prostaglandins, and mechanical stimuli, such as fluid shear stress (FSS). Although they have been less investigated, mechanical stimuli have a fundamental role in modulating the delicate equilibrium between endothelial function and dysfunction. FSS varies greatly based on the magnitude, direction, and flow speed. Accordingly, FSS can be distinguished in steady laminar, disturbed laminar, oscillatory, and turbulent [37][67]. Endothelial cells are critical sensors of FSS, and they undergo morpho-functional modifications as a physiological response. However, turbulent FSS can stimulate pathological endothelial remodeling, leading to an increased risk of developing cardiovascular diseases, such as atherosclerotic plaque formation and hypertension [38][68]. Aging is associated with significant structural and mechanical alterations of vascular endothelium, including the increase of intimal-to-media thickness, aortic length and circumference, and stiffness as well as the reduction of elasticity and distensibility and endothelial capacity to respond to mechanical stresses. All these mechanisms contribute to age-related hypertension. Chala et al., in an in vitro model of endothelial senescence, showed that endothelial cells have a reduced capacity to adapt to the local hemodynamic conditions [39][69] functionally. Interestingly, recent experimental studies showed that disturbed flow induces endothelial senescence. Thus, vascular FSS could be a critical determinant in regulating vascular senescence [40][70].Causative Mechanisms of Endothelial Alterations in Hypertension
Several molecular pathways underpinning the structural and functional endothelial alterations have been described in age-related hypertension, including Sirtuins, oxidative stress, and the renin-angiotensin-aldosterone system (RAAS). Sirtuins are a family of proteins with mono-ADP-ribosyltransferase and deacetylase activity, initially described as class III histone deacetylases [41][71]. It consists of seven members with different cellular localization and functions. Sirtuins have a protective effect against the decline in cell function. Specifically, SIRT1 is highly expressed in the endothelial cells of arteries, veins, and capillaries, and plays a critical role in preventing endothelial dysfunction [42][72]. It has been shown that the SIRT1 gene is downregulated during aging. In vivo and in vitro studies showed that the overexpression of SIRT1 slows down endothelial senescence [43][73]. As stated above, oxidative stress is a hallmark of aging and plays a prominent role in orchestrating age-related endothelial dysfunction. The primary source of ROS is mitochondrial oxidases, NADPH oxidases (NOX), and eNOS. Data from in vivo and in vitro indicate a leading role of NOX [44][45][74,75]. NOX is a family of transmembrane enzymes consisting of seven members. Among these, Nox1, Nox2, Nox4, and Nox5 are mainly expressed in the vascular system. The increase in NOX activity has been described in the experimental model of ageing [46][76]. Noteworthy, the age-dependent increase of NOX is accompanied by the alteration of several molecular pathways, including the improved activation of RAAS, which represents a key regulator of blood pressure. Despite their undiscussed importance in physiological and pathological vascular function, a few studies explored mechanistic insight into the role of NOX in aging. Age-related oxidative stress results from the imbalance between ROS production and degradation. Indeed, aging promotes ROS production while inhibiting antioxidant systems with the net increase of ROS levels. ROS stimulates cellular pathways, leading to aberrant cell signaling and post-translation modifications, such as phosphorylation and oxidation, which impair fundamental cellular processes [47][77]. The reversible post-translational modifications are critical for regulating protein function (activation or inactivation). Specifically, ROS activate molecular pathways, leading to vascular remodeling, inflammation, and hypertension. Additionally, ROS can block the Sirt1 function and reduce NO concentration by increased quenching and eNOS inhibition. Overall, all these mechanisms contribute to endothelial dysfunction and related hypertension. A mutualistic relationship between oxidative stress and RAAS has been described [48][78]. RAAS is the critical regulator of blood pressure and consists of three main components: renin, angiotensin II, and aldosterone [49][79]. Furthermore, in vitro studies showed that angiotensin II stimulates the production of superoxide anions, while the inhibition of RAAS is associated with a reduction of oxidative stress [50][80]. Thus, robust evidence supports a strict relationship between RAAS and oxidative stress, underpinning age-related hypertension. In the last decade, a role for endothelial progenitor cells (EPC) has emerged [51][81]. EPCs are a heterogeneous cellular population originating and residing in different organs and tissues, including vascular endothelium. After an injury, they are involved in vascular remodeling by replacing the dysfunctional endothelial cells. It has been shown that aging and hypertension are independently associated with reducing EPCs [52][82]. Furthermore, cumulating literature evidence shows that oxidative stress induces the reduction of EPCs number. Thus, the interplay among oxidative stress reduced NO bioavailability and decreased EPCs number creates a vicious circle, boosting endothelial dysfunction. Beyond the well-known role of hypertension in the increased risk of stroke, coronary heart disease, sudden death, heart failure, and peripheral artery disease, age-related hypertension predisposes the onset of cognitive and functional decline, resulting in dementia and physical frailty, respectively. It should be noted that hypertension is commonly underdiagnosed in patients older than 80 years. Indeed, the evaluation of such clinical conditions in the elderly is hampered by several factors, including the poor compliance of the patient and the presence of multiple comorbidities which could hide hypertension [53][83]. Thus, there is an urgent need to appropriately manage hypertension in the elderly, from diagnosis to treatment.2.2. Endothelial Dysfunction in Diabetes
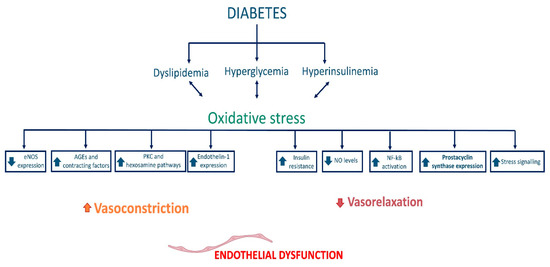
Type 2 diabetes mellitus (T2D) is a metabolic disease characterized by reduced insulin sensitivity by target organs or reduced insulin production by pancreatic beta cells. Whatever the initial pathophysiologic features, the reduction in insulin sensitivity leads over time to an increase in insulin production to cope, and subsequently to depletion and reduction in pancreatic beta-cell stock due to exhaustion, resulting in hyperglycemia. Certainly, given the pleiotropic effects of insulin, because of the altered glucose metabolism and the effects on lipid metabolism (insulin is an anabolic hormone), T2D is often associated with obesity. However, this association is not always present: there is the nondiabetic obese patient, and there are diabetic subjects who remain at an average weight throughout their life [54][84]. Undoubtedly, aging promotes the onset of metabolic disorders, and this is the fundamental reason why both T2D and obesity are more frequent in the aging population and why the two diseases often occur together: the senescence that affects other cells in the body does not spare pancreatic beta cells, which become less efficient and more likely to fail as the time passes. In addition to this, several studies have documented the presence of senescent beta cells in the pancreatic islets of donors with T2D and subjects with high BMI, further demonstrating that several factors, including dysmetabolism, play a crucial role in aging [55][85]. Hyperglycemia constitutes a harmful condition for the homeostasis of cells, including endothelial cells, and consequently represents the characteristic trigger of diabetes on vascular pathologies. In addition, high serum glucose levels cause functional and metabolic changes through reduced nitric oxide bioavailability, increased oxidative stress, and induction and acceleration of cellular senescence through direct action on the NF-κB pathway. Under hyperglycemic conditions, the induction of transcription of the gene for the NF-κB factor appears to depend on the activation of molecules of the guanosine triphosphate (GTP)-ases family, which in turn promotes the induction of transcription of inflammatory mediator genes such as IL-1 β, TNF-α, protein kinase C, and adhesion molecules of the cellular adhesion molecule (CAM) family, which facilitate the binding of circulating cells (namely monocytes and T lymphocytes) to endothelial cells. Atherosclerosis in the diabetic subject is therefore strongly catalyzed by hyperglycemia: on the one hand, inflammation worsens plaque formation as it encourages the oxidation of low-density lipoproteins, and on the other, the reduction in nitric oxide bioavailability, due to sustained endothelial damage, causes hemodynamic alterations and increased shear stress, which are the foundations of vascular pathologies. In healthy subjects, vasoregulation results from a delicate balance between the vasoactive substances produced by the endothelium and the shear forces exerted by the blood flow. Thanks to continuous feedback, the mechanotransduction of these shear forces allows the endothelium to produce vasodilating substances such as nitric oxide or prostacyclin (PGI2), or vasoconstrictive substances such as endothelin (ET) as required. Sulistyowati et al. documented that hyperglycemia impairs this mechanism, as it disrupts the functioning of endothelial cells by reducing endothelial nitric oxide synthase (eNOS) activity and disrupting mechanotransduction activity, generating the hemodynamic abnormalities discussed above [56][86]. Concerning nitric oxide, its production in endothelial cells depends on the phosphorylation of the enzyme eNOS by serine/threonine kinases acting coordinately in the insulin signaling pathway; hyperglycemia alters the binding and activation of the insulin receptor, thus leading to an impairment of this molecular mechanism and a reduction of nitroxide [57][87]. Hyperglycemia impairs endothelial function through a variety of mechanisms: through induction of advanced glycation end products (AGEs) formation, increased expression of receptors for AGEs (RAGE), increased polyol production, and hyperactivation of the hexosamine pathway [58][88]. The genesis of AGEs causes structural and functional alterations and results in receptor recognition abnormalities of matrix components. In addition, the interaction between ligand (AGE) and its receptor (RAGE) results in increased superoxide anion production that promotes macrophage-mediated inflammatory vascular damage [59][60][89,90]. Moreover, the increase in AGEs in the bloodstream reduces nitric oxide synthesis by endothelial cells by reducing the expression of the enzyme eNOS and, consequently, the bioavailability of this vasodilation mediator, conversely facilitating the synthesis of endothelin-1, which mediates vasoconstriction (Figure 1) [61][62][91,92].