Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Omar Soler-Cedeño | -- | 3734 | 2023-01-19 20:22:59 | | | |
| 2 | Rita Xu | Meta information modification | 3734 | 2023-01-20 02:17:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Soler-Cedeno, O.; Xi, Z. Neutral CB1 Receptor Antagonists. Encyclopedia. Available online: https://encyclopedia.pub/entry/40425 (accessed on 23 July 2026).
Soler-Cedeno O, Xi Z. Neutral CB1 Receptor Antagonists. Encyclopedia. Available at: https://encyclopedia.pub/entry/40425. Accessed July 23, 2026.
Soler-Cedeno, Omar, Zheng-Xiong Xi. "Neutral CB1 Receptor Antagonists" Encyclopedia, https://encyclopedia.pub/entry/40425 (accessed July 23, 2026).
Soler-Cedeno, O., & Xi, Z. (2023, January 19). Neutral CB1 Receptor Antagonists. In Encyclopedia. https://encyclopedia.pub/entry/40425
Soler-Cedeno, Omar and Zheng-Xiong Xi. "Neutral CB1 Receptor Antagonists." Encyclopedia. Web. 19 January, 2023.
Copy Citation
Cannabinoid receptor 1 (CB1R) has been one of the major targets in medication development for treating substance use disorders (SUDs). Early studies indicated that rimonabant, a selective CB1R antagonist with an inverse agonist profile, was highly promising as a therapeutic for SUDs.
cannabinoid
CB1 receptor
Δ9-tetrahydrocannabinol
rimonabant
substance use disorders
1. Introduction
Substance use disorder (SUD), defined as the uncontrollable and persistent use of drugs (including alcohol) despite substantial harm and adverse consequences, is still a severe social and health problem worldwide. SUD-related costs, including those in crimes, loss of productivity and healthcare, exceed $740 billion per year in the Unites States [1]. In recent years, opioid overdose and SUD-related diseases have increased dramatically with the fatal incidents up to ~50,000 in 2017 in the USA [2]. Although the United States Food and Drug Administration (FDA) approved several medications such as methadone, buprenorphine, and varenicline for the treatment of opioid or nicotine use disorders [3][4][5], the rate of relapse remains extremely high. Moreover, there is no FDA-approved medication for the treatment of psychostimulant use disorders [6]. Over the past decades, the cannabinoid receptor 1 (CB1R) has been given much attention as a promising target in medication development for treating SUDs [7][8][9]. The reason for such attention is because of convincing evidence indicating that rimonabant, a selective CB1R antagonist with an inverse agonist profile, is highly effective in reducing drug taking and drug-seeking behavior in experimental animals [7][8][10]. However, the severe adverse effects of rimonabant, such as nausea, emesis, depression, and suicidal tendencies observed in humans have led to its withdrawal from clinical trials worldwide [11]. Consequently, the US FDA decided not to approve CB1R ligands until better safety and efficacy data become available.
2. Mesocorticolimbic Dopamine System
To better understand how cannabinoid CB1R antagonists produce anti-addictive effects, it is necessary to briefly review the current working hypothesis underlying drug reward and addiction. Addiction includes three stages—binge/intoxication, a stage at which an individual consumes an intoxicating substance and experiences its rewarding effects; withdrawal/negative affect, a stage at which an individual experiences a negative emotional state in the absence of the substance; and preoccupation/anticipation, a stage at which subject seeks substances again after a period of abstinence [12]. Although the understanding of the neural mechanisms underlying each stage of addiction is still not fully understood, a well-accepted view is that the rewarding effects of drugs of abuse are mediated mainly by activation of the mesocorticolimbic dopamine (DA) system. This system originates in DA neurons in the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc) of the midbrain and projects to the prefrontal cortex (PFC), nucleus accumbens (NAc), and the dorsal striatum (SD) [6][13]. Different drugs of abuse activate this pathway by distinct receptor and cellular mechanisms [14][15][16] (Figure 1). For example, the psychostimulant cocaine activates this system mainly by blocking the DA transporter (DAT), while nicotine activates VTA DA neurons by stimulating nicotinic receptors located on DA neurons or glutamate neurons that project to DA neurons in the VTA and NAc [16][17][18]. Alcohol’s reinforcement has been associated with processes involving multiple molecular targets, including mu opioid receptors and NMDA receptors [12][19][20][21]. On the other hand, opioids activate midbrain DA neurons mainly by stimulation of opioid receptors located on GABAergic neurons in the rostromedial tegmentum (RMTg) and substantia nigra pars reticulata (SNr) that project to the VTA and SNc, respectively, causing increases in DA neurons firing and striatal DA release via GABA-mediated disinhibition [5][17][22]. Therefore, both the RMTg-VTA-NAc and SNr-SNc-DS DA pathways play a central role in drug reward and addiction [14][22], making the DA system a crucial target in medication development for the treatment of SUDs.
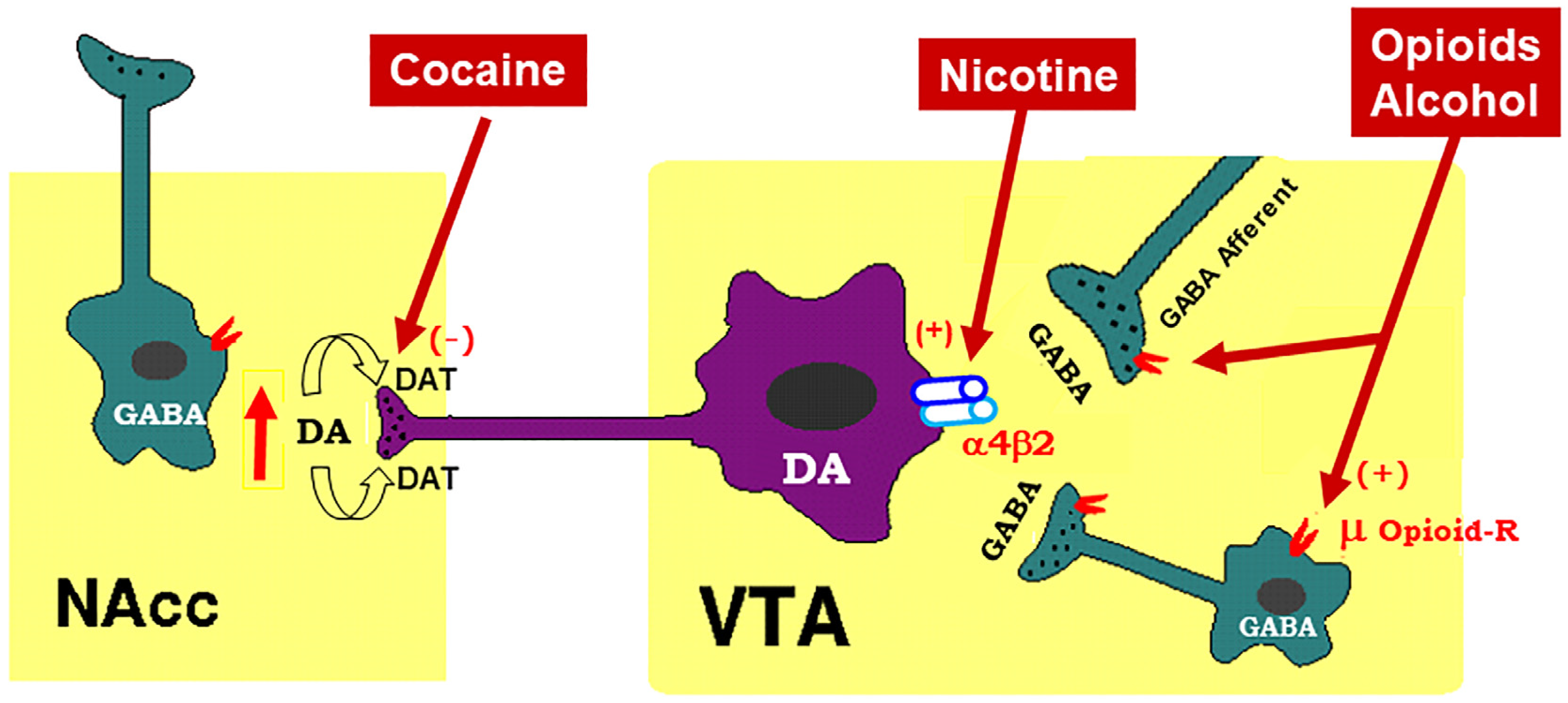
Figure 1. Schematic diagram of the mesolimbic dopamine (DA) hypothesis, illustrating how drugs of abuse activate this system. The mesolimbic DA system originates in the midbrain ventral tegmental area (VTA) and projects predominantly to the forebrain nucleus accumbens (NAc) and the prefrontal cortex (not shown). The psychostimulant cocaine elevates extracellular NAc DA by blocking DA transporters (DAT) on DA axon terminals, while opioids (such as heroin) and alcohol bind to and activate mu opioid receptor (μ Opioid–R) located mainly on GABAergic afferents (less on VTA GABAergic interneurons) and inhibit GABA release. A reduction in GABA release leads to DA neuron disinhibition (activation). Nicotine has been thought to activate DA neurons mainly by activation of α4β2 nicotinic receptors located on DA neurons. (+), (-): indicate activation of opiate receptors or blockade of DAT.
3. Endocannabinoid System
To better understand how CB1R antagonists produce therapeutic effects against drug abuse and addiction and how CB1R inverse agonists produce unwanted side-effects, let researchers briefly review the endocannabinoid (eCB) system and recent research on how cannabinoids modulate the mesocorticolimbic DA system, a critical action site for drugs of abuse.
The endocannabinoid (eCB) system consists of cannabinoid receptors (CB1Rs, CB2Rs, and others), endocannabinoids [anandamide (AEA) and 2-arachidonoylglycerol (2-AG)], enzymes for endocannabinoid synthesis [N-arachidonoyl phosphatidylethanolamine-phospholipase D (NAPE-PLD), diacylglycerol-lipase (DAG-lipase)] and degradation [fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MGL)], and their putative transport systems [23][24]. AEA was the first endocannabinoid discovered by Raphael Mechoulam and his colleagues in 1992 [25]. AEA is an endogenous CB1R agonist (Ki = 87.7 nM for rCB1; Ki = 239.2 nM for hCB1) and a weak CB2R agonist (Ki = 267.8 nM for rCB2; Ki = 439.5 nM for hCB2) [26]. The effects of AEA are mediated mainly by activation of CB1Rs and CB2Rs in the brain and periphery. However, AEA levels in the brain are very low in healthy subjects, and it has a very short half-life (~2 min) due to its fast degradation by fatty acid amide hydroxylase (FAAH) [27]. Therefore, the functional significance of AEA in the brain is largely unclear.
2-AG was the second eCB discovered in the brain [28]. It is an endogenous agonist of the CB1Rs (Ki = 1180 nM for rCB1; Ki = 3423 nM for hCB1) and CB2Rs (Ki = 1900 nM for rCB2; Ki = 1193 nM for hCB2) [26]. Unlike AEA, 2-AG is present at relatively high levels in the central nervous system (CNS). Therefore, it is thought to be a major eCB modulating brain function.
There are at least two types of cannabinoid receptors (CB1Rs and CB2Rs) identified in the brain [23]. The phytocannabinoids (Δ9-THC), synthetic cannabinoids (WIN55,212-2, CP55,940, HU-210), and the endocannabinoids (AEA, 2-AG) all have high binding affinities at both the CB1Rs and CB2Rs [23]. Cannabinoids may also bind to other putative cannabinoid receptors, such as G protein-coupled receptor 55 (GPR55), transient receptor potential vanilloid 1 (TRPV1) channel, and peroxisome proliferator-activated nuclear receptors (PPARs) [23]. Accumulative evidence indicates that cannabinoid action is mediated mainly by activation of CB1Rs and CB2Rs [23].
4. Cannabinoid Reward versus Aversion
Cannabis is the most commonly used substance worldwide as many people find it pleasurable [29]. However, the findings regarding the rewarding properties of cannabinoids in both humans and experimental animals are conflicting [23]. Indeed, cannabis use has often been associated with its psychoactive, rewarding effects [30][31]. The psychoactive effects of cannabis, combined with the ongoing cannabis legalization in the United States, may well explain why cannabis use is rising in the USA. For instance, from 2002 to 2019, the percentage of adults who reported using cannabis in the past year increased from 7.0 to 15.2% [32].
However, cannabis enjoyment is not universal, and some individuals report dysphoria, anxiety, and depression after cannabis use [33][34]. The increase in cannabis use also raises concerns about possible adverse effects of cannabis use, such as developing the amotivational syndrome [35][36], which is defined as “a reduction in the motivation to initiate or persist in goal-directed behavior” [37]. A series of human functional magnetic resonance imaging (fMRI) studies support these cannabis amotivational effects by evidence that Δ9-THC produces a significant reduction in reward-related brain activity or neural response to reward in healthy adults [38][39][40]. In congruent with these findings, other reports showed that Δ9-THC reduced the likelihood or motivation of reward-related learning and decision-making [41], dampened neural responses to music [42], and reduced striatal DA response to reward [43].
Similar paradoxical effects of cannabinoids have been discovered in non-human primates, as squirrel monkeys self-administer Δ9-THC or endocannabinoids [44][45], while other primate species (rhesus, baboon, cynomolgus) fail to demonstrate this behavior [46][47][48]. In rodents, Δ9-THC alone is not self-administered [49][50], although the mixture of Δ9-THC and cannabidiol was recently reported to be self-administered by rats [51][52]. In conditioned place preference (CPP) test, Δ9-THC typically produces conditioned place aversion [53][54], although place preferences have also been reported [55][56]. In electrical intracranial self-stimulation (ICSS) experiments, Δ9-THC was initially reported to facilitate electrical ICSS in rats [56][57][58], while other studies found suppression of ICSS in rats and mice [59][60][61][62][63]. In optogenetic ICSS (oICSS) maintained by optical stimulation of midbrain DA neurons or glutamate neurons, cannabinoids always produce a reduction in brain-stimulation reward (BSR) in mice, suggesting a reward-attenuating or aversive effect [64][65].
Similarly, the findings of cannabinoid action on DA transmission are also conflicting. There are reports indicating that activation of the CB1Rs increases DA neuronal firing in the VTA [66][67] and DA release in the NAc in rats [68][69][70][71]. However, in vitro voltammetry experiments in striatal brain slices demonstrate that the cannabinoids WIN55,212-2 or CP55,940 fail to alter [72][73] or produce a reduction in electrical stimulation-induced DA release in the dorsal striatum in guinea pigs, rats and mice [74][75]. In vivo microdialysis experiments in freely moving animals indicate that Δ9-THC produces a dose-dependent reduction in NAc DA in mice [76]. The neural mechanisms underlying such opposite affective and neurochemical effects of cannabinoids are not fully understood.
4.1. GABAergic CB1R Hypothesis of Cannabis Reward
Given that midbrain DA neurons receive both inhibitory GABAergic and excitatory glutamatergic inputs, researchers proposed that differential CB1R expression on GABAergic neurons versus glutamatergic neurons may underlie cannabinoid reward versus aversion, respectively [7][23][64] (Figure 2). The GABAergic CB1R hypothesis is supported by electrophysiological findings in brain slices where stimulation of CB1Rs on VTA GABAergic neurons causes an increase in VTA DA neuron firing via GABA-mediated disinhibition [77][78][79][80]. However, so far, there is a lack of behavioral evidence in vivo supporting this GABA-CB1R hypothesis possibly due to the absence of reliable behavioral models of cannabinoid reward in rodents.
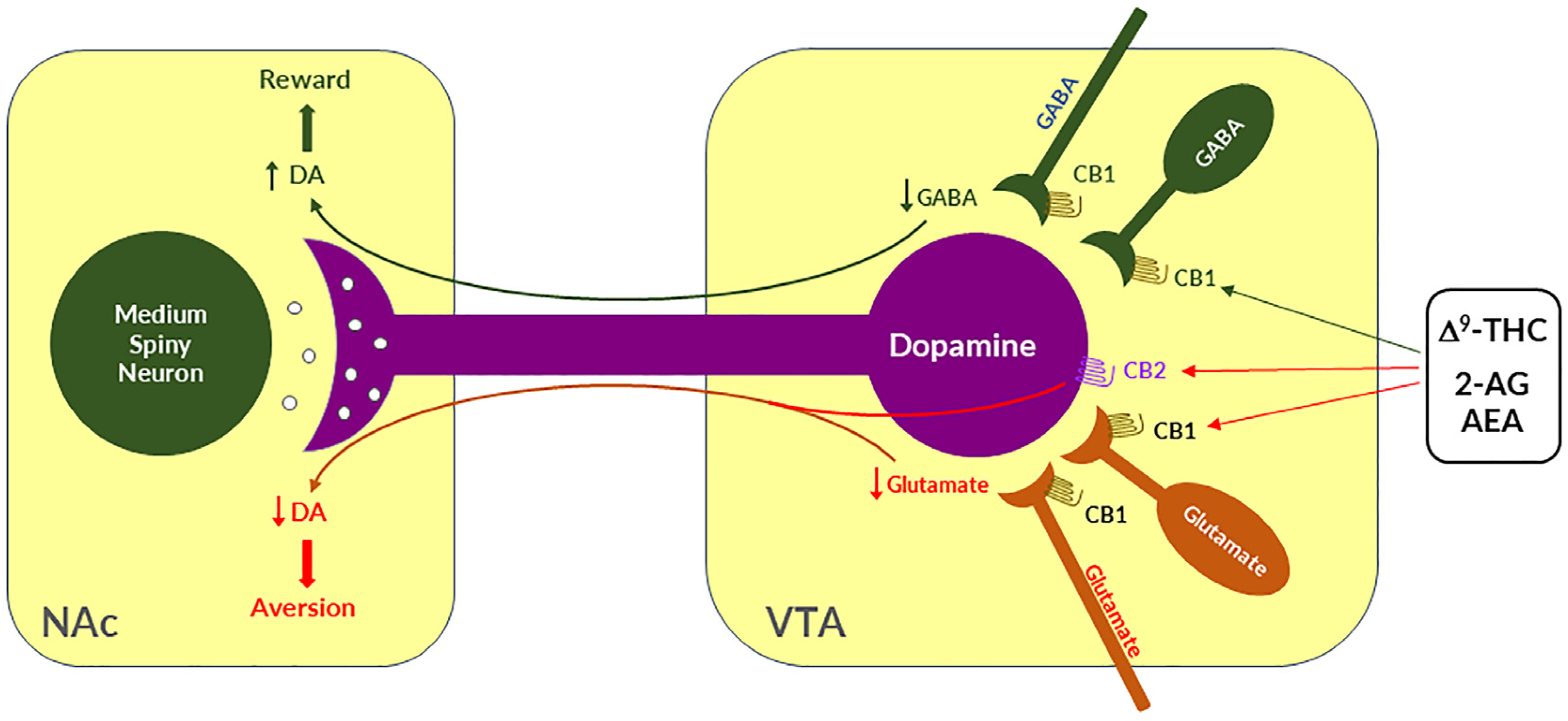
Figure 2. A schematic diagram showing the neural mechanisms underlying cannabis reward versus aversion. Cannabinoid CB1Rs are expressed not only in VTA GABAergic interneurons or GABAergic afferents but also in VTA glutamatergic neurons or their afferents within the VTA, whereas CB2Rs are also expressed in VTA DA neurons. Both CB1Rs and CB2Rs are inhibitory G-protein (Gi)-coupled receptors, producing an inhibitory effect on neuronal firing or terminal neurotransmitter release after activation. Cannabinoids such as Δ9-THC, 2-AG, and AEA may produce rewarding effects by binding to CB1Rs on VTA GABAergic interneurons and/or their afferents as a reduction in GABA release causes an increase DA neuronal firing and enhanced DA release in the NAc. Conversely, cannabinoids may also produce aversive effects by activating CB1Rs on glutamatergic neurons and/or terminals in the VTA that decreases excitatory glutamate input on VTA DA neurons. In addition, activation of CB2Rs on VTA DA neurons also produce an inhibitory effect on DA neuron firing and DA release in the NAc. Thus, the subjective effects of cannabinoids may depend on the balance of both oppose actions. This hypothesis may well explain why cannabinoids are rewarding in some subjects or species, while ineffective or even aversive in others. ↑, ↓—indicate an increase or a decrease in neurotransmitter release.
4.2. Glutamatergic CB1 Hypothesis of Cannabinoid Aversion
Clearly, the above GABAergic disinhibition hypothesis cannot explain how cannabinoids produce the aversive effects observed in rodents. To address this question, researchers have recently used advanced RNAscope in situ hybridization (ISH) assays to examine the cellular distributions of CB1Rs. They found that CB1Rs are expressed not only in VTA GABAergic neurons but also in VTA glutamatergic neurons [23][64][65]. Strikingly, optogenetic activation of VTA glutamatergic neurons produced potent rewarding effects, as assessed by CPP and optical ICSS (oICSS) [64][81]. Systemic administration of multiple cannabinoids (such as Δ9-THC, WIN55,212-2, ACEA, AM-2201) dose-dependently inhibited glutamate-mediated oICSS only in VgluT2-Cre control mice, but not in glutamate-CB1-knockout mice in which CB1Rs are selectively deleted from subcortical VgluT2-expressing glutamate neurons [64]. These findings suggest that activation of CB1Rs on glutamate neurons produces reward-attenuation or aversive effects by decreasing glutamatergic inputs onto VTA DA neurons (Figure 2).
The above findings also suggest that activation of brain CB1Rs is not always rewarding but it could be aversive, depending upon the cellular distribution of CB1R expression in the brain. Therefore, researchers propose that the hedonic effects of cannabis might depend on the balance of two opposing actions of cannabinoids on both GABAergic neurons and glutamatergic neurons (Figure 2). If more CB1Rs are expressed in VTA or VTA-projecting GABAergic neurons, cannabis will be rewarding as GABAergic disinhibition of VTA DA neurons is dominant. In contrast, if more CB1Rs are expressed in VTA or VTA-projecting glutamatergic neurons, cannabis will be aversive as CB1R mediated reduction in glutamatergic inputs onto VTA DA neurons is dominant. Congruently, if CB1R levels are equivalent on both types of neurons, cannabis should have no net effect on the brain reward function (Figure 2). This hypothesis appears to well explain why Δ9-THC or cannabinoids are rewarding in some human subjects and non-human primates (squirrel monkeys) in which more CB1Rs might be expressed in VTA-projecting GABA neurons but are ineffective or aversive in other human subjects and species (such as rats, mice) in which more CB1Rs might be expressed in VTA or VTA-projecting glutamate neurons (Figure 2).
4.3. Dopaminergic CB2 Hypothesis of Cannabinoid Aversion
In addition to the above glutamate-CB1R hypothesis, recent research indicates that CB2Rs are also expressed in VTA DA neurons and contribute to the aversive effects of cannabinoids [3][82]. Activation of CB2Rs by JWH133 inhibits VTA DA neuron activity and decreases NAc DA release in wildtype mice, but not in CB2-KO mice [83][84][85][86]. Activation of CB2Rs or overexpression of brain CB2Rs also inhibits cocaine self-administration, cocaine-induced CPP and hyperactivity in mice [85][87][88][89]. In rats, Δ9-THC and WIN55, 212-2 produce biphasic effects on electrical brain-stimulation reward (BSR)—reward-enhancing at lower doses and reward-attenuating (or aversive) at higher doses [63]. CB1R antagonism (by AM251) reduced the low dose-enhanced BSR, while CB2R antagonism (AM630) decreased the high dose-attenuated BSR. Congruently, selective CB1R and CB2R agonists produced significant BSR enhancement and inhibition, respectively [63]. Together, these findings suggest that DA-CB2R mechanisms, at least in part, underlie cannabinoid-induced aversion (Figure 2) [3][23]. Thus, the subjective effects of cannabinoids would depend on the balance of multiple cell type-specific receptor mechanisms. This cell type-specific cannabinoid receptor mechanism appears to well explain why cannabis or cannabinoids could be rewarding, aversive, or ineffective since the cellular distributions of CB1Rs and CB2Rs may be different in different subjects or species.
5. CB1R Antagonists Are Promising for the Treatments of SUDs
Rationale: In a series of clinical trials known as the Studies with Rimonabant and Tobacco Use (STRATUS), it was found that rimonabant significantly increased abstinence rates and reduced smoking cessation-related weight gain [9][90][91][92][93], suggesting that the endocannabinoid system may also be involved in nicotine use disorder and CB1R antagonists or inverse agonists may be useful for the treatment of SUDs including nicotine use disorder.
Supporting evidence: The findings with either CB1R agonists or antagonists support this hypothesis of eCB involvement in SUDs. For example, Δ9-THC increases heroin self-administration in rats [94]. WIN55,212-2 increases motivation to nicotine self-administration and facilitates cue-induced reinstatement of nicotine seeking in rats [95]. WIN55,212-2 or CP55,940 facilitate alcohol self-administration, CPP, and binge-like behavior in rodents [96][97]. Accordingly, it was proposed that CB1R antagonists should also be effective in the treatment of SUDs [8][98][99].
Indeed, compelling preclinical evidence supports this hypothesis. For example, rimonabant reduced intravenous heroin self-administration under fixed-ratio and progressive-ratio reinforcement [100][101], nicotine cue-induced reinstatement of nicotine-seeking behavior in rats [102][103], and nicotine-enhanced DA release in the NAc [102]. Furthermore, rimonabant also prevents the development of morphine-induced CPP [104], and dose-dependently attenuates heroin- or heroin-associated cue-induced reinstatement of drug-seeking behavior [101]. In addition, rimonabant blocks acquisition of cocaine-induced CPP [105] and attenuates reinstatement of drug seeking caused by cocaine or cocaine-associated cues [106]). Congruently, rimonabant also blocks reinstatement of methamphetamine-seeking behavior [107] and reduces alcohol intake in rodents [108]. These exciting findings with rimonabant encouraged many pharmaceutical industries to develop other brain-penetrant CB1R antagonists such as the longer acting second generation surinabant (SR147778, Sanofi-Aventis), taranabant (MK-0364, Merck), otenabant (CP-945,598, Pfizer), and ibipinabant (SLV319, Solvay Pharmaceutical), for the treatment of obesity, smoking and drugs of abuse [109].
Receptor mechanisms: Unfortunately, all the above-mentioned ligands are not only CB1R antagonists but also inverse agonists. Thus, dissecting the role of CB1R antagonism versus inverse agonism in their therapeutic versus side-effects is critical for developing safer CB1R ligands for the treatment of SUDs.
CB1R antagonism may underlie the therapeutic effects of rimonabant: Given that rimonabant is a well-characterized CB1R antagonist, it is reasonable to hypothesize that the CB1R antagonism may underlie its therapeutic anti-obesity and anti-addictive effects. This is supported by several lines of evidence. First, cannabinoids and drugs of abuse may act in a common neural substrate—the mesocorticolimbic DA system via distinct cellular and receptor mechanisms (Figure 2). Assuming that CB1Rs are tonically activated by eCBs (2-AG, AEA), CB1R antagonism on GABA neurons or GABAergic terminals would produce a reduction in eCB-enhanced DA transmission (Figure 2), which may functionally counteract the DA-enhancing and the pro-addictive effects produced by drugs of abuse. Second, growing evidence indicates that drugs of abuse (such as cocaine, heroin, or nicotine) may increase eCB release in the VTA and/or NAc [110][111][112][113][114][115][116][117] (Figure 3). After releasing from post-synaptic neurons, such as VTA DA neurons (Wang et al., 2015), eCBs retrogradely diffuse back to activate presynaptic CB1Rs, producing a reduction in neurotransmitter (GABA, glutamate) release, which subsequently produces reward-enhancing and addictive effects (Figure 3). Accordingly, CB1R antagonism at presynaptic terminals would block the eCB-mediated effects, producing anti-addictive effects (Figure 3). Third, the neutral CB1R antagonists without CB1R inverse agonist profile (such as AM4113 and PIMSR) produce the similar anti-addictive effects as rimonabant in drug self-administration and reinstatement, but without rimonabant-like depressive effects [7][13][118]. Lastly, in theory, rimonabant’s anhedonic or aversive effects may also functionally counteract the rewarding effects of drug abuse; however, direct supporting evidence is missing due to the absence of selective CB1R inverse agonists.
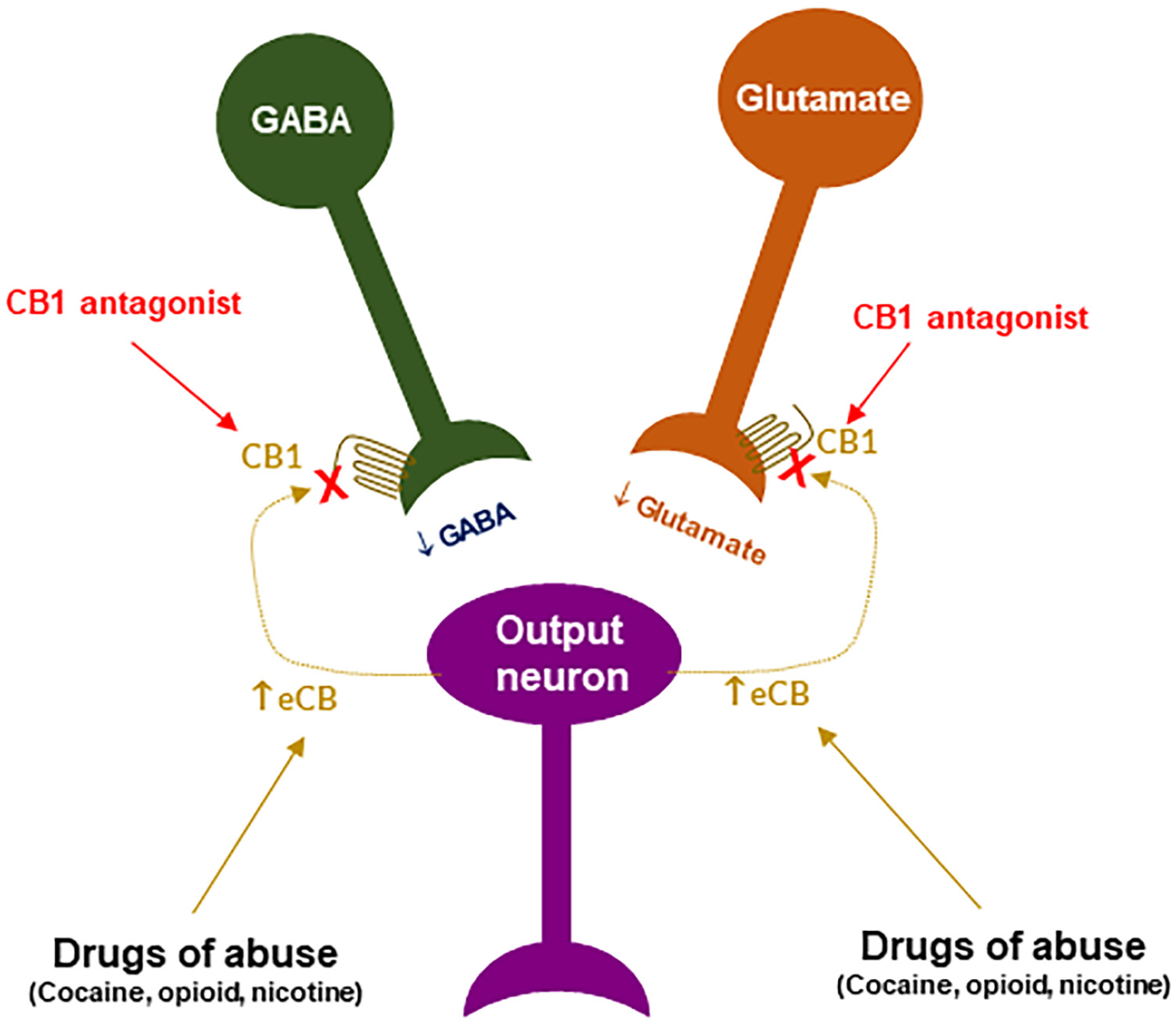
Figure 3. A schematic diagram of an endocannabinoid (eCB) hypothesis showing how a CB1R antagonist blocks actions produced by drugs of abuse. Growing evidence indicates that drugs of abuse (such as cocaine, opioid or nicotine) may stimulate endocannabinoid (such as 2-AG, AEA) release from postsynaptic neurons (such as VTA DA neurons), which subsequently activates CB1R on presynaptic CB1Rs and produces a reduction in GABA or glutamate release. A CB1R antagonist, including neutral antagonist, binds to CB1R and blocks eCB binding to the same receptor, therefore producing antagonism of drug reward and relapse. ↑: indicates an increase in eCB release; “×” indicates blockade of CB1 receptor.
CB1R inverse agonism may underlie the adverse effects of rimonabant: As discussed above, CB1R agonism by Δ9-THC on GABA neurons or GABAergic terminals may produce an increase in NAc DA release and reward-enhancing effects via a disinhibition mechanism (Figure 2 and Figure 4A). Accordingly, CB1R inverse activation by rimonabant on the same receptor would produce opposite effects—enhanced GABA and reduced NAc DA release, which may translate into depressive-like subjective effects of rimonabant (Figure 4B).
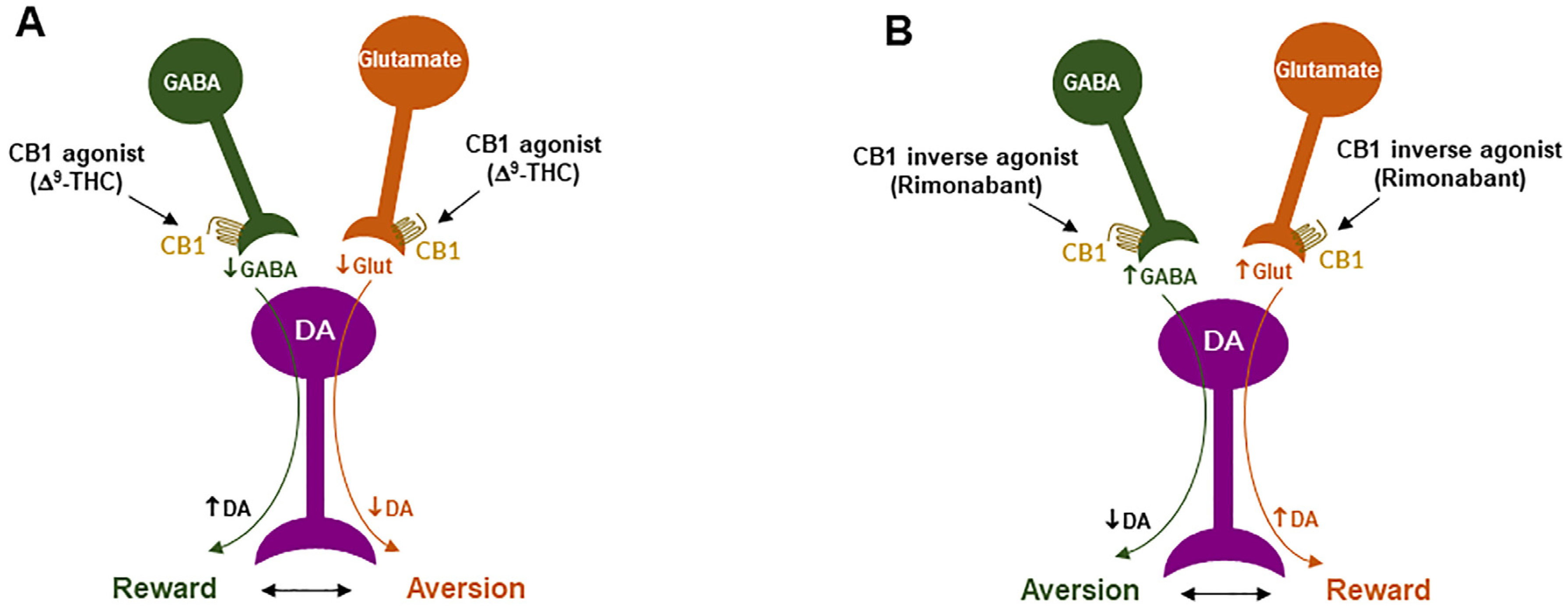
Figure 4. Schematic diagrams of a working hypothesis showing how a CB1R agonist and an inverse agonist produce opposite subjective effects. (A): A CB1R agonist such as Δ9-THC may produce rewarding effects by decreasing VTA GABA release and increasing NAc DA release. (B): In contrast, an inverse CB1R agonist may produce an opposite aversive effect by increasing GABA release and decreasing DA release in the NAc. ↑, ↓: indicate an increase or a decrease in neurotransmitter release.
Three animal models are often used to evaluate the rewarding versus aversive effects [119][120]. They are intracranial self-stimulation (ICSS) (also called brain-stimulation reward, BSR), NAc DA response to test drugs, and conditioned place preference/aversion (CPP/CPA). The findings with rimonabant from these models are mixed (Table 1). In the ICSS model, researchers [118] and others [121] previously reported that high-dose rimonabant inhibits electrical ICSS, while three other reports [122][123][124] indicate that rimonabant has no effect on ICSS. With respect to DA response to rimonabant, researchers have previously reported that, in mice, systemic administration of rimonabant produces a reduction in extracellular DA in the NAc [125]. However, in Long-Evan rats, researchers found that systemic rimonabant failed to alter extracellular NAc DA, while intra-NAc local perfusion of rimonabant at 100 µM (but not at 0.1, 1.0, or 10.0 µM) unexpectedly increased extracellular NAc DA [76]. Another report indicates that—in Wistar rats—rimonabant produced enhanced NAc DA response [126]. In the CPP/CPA model, most publications reported that rimonabant, at low doses, produced neither CPP nor CPA (Table 1). Thus, although some evidence supports that rimonabant could be aversive by itself, which could be related to its inverse agonist profile, conclusive supporting evidence is still lacking. The negative findings may be related to rimonabant doses tested in the above studies and/or the distinct cellular distributions of CB1R expression in different species (rats vs. mice) or subjects under different experimental conditions. As stated below, the important findings with PIMSR, a neutral CB1R antagonists without inverse agonist profile, in the same animal models provide valuable evidence supporting an assumption that the CB1R inverse agonism at least in part underlie the adverse psychiatric effects of rimonabant, as discussed above.
Table 1. The literature reports regarding the rewarding vs. aversive effects of SR141716 itself in rodents.
| Compound | Doses | Species | Results | References |
|---|---|---|---|---|
| Rimonabant | 20 mg/kg, i.p. | Rat | ↓ Electrical brain-stimulation reward | [121] |
| Rimonabant | 0.3, 1, 3, 10 mg/kg, i.p. | Rat | ↓ Electrical brain-stimulation reward | [118] |
| Rimonabant | 0.02, 0.3, 1.0 mg/kg, i.p. | Rat | No effect on electrical brain-stimulation reward | [123] |
| Rimonabant | 0.02 mg/kg, i.p. | Rat | No effect on electrical brain-stimulation reward | [124] |
| Rimonabant | 3, 10 mg/kg, i.p., | Mouse | No effect on electrical brain-stimulation reward | [122] |
| Rimonabant | 0.3, 1, 3 mg/kg, i.p. | Rat | Not produce CPP or CPA | [105] |
| Rimonabant | 0.1, 0.5, 3.0 mg/kg, i.p. | Rat | Not produces CPP or CPA | [127] |
| Rimonabant | 0.5, 1, 2 mg/kg, i.p. | Rat | Not produce CPP or CPA | [128] |
| Rimonabant | 3 mg/kg, i.p. | Rat | Not produce CPP or CPA | [89] |
| Rimonabant | 0.25, 0.5, 1 mg/kg, i.p. | Rat | Not produce CPP or CPA | [55] |
| Rimonabant | 0.1, 0.5, 3 mg/kg | Rat | Not produce CPP or CPA | [129] |
| Rimonabant | 0.3, 3 mg/kg, i.p. | Rat | Not produce CPP or CPA | [130] |
| Rimonabant | 0.25, 0.5, 2, 3 mg/kg | Rat | Produces CPP | [53] |
| Rimonabant | 3 mg/kg | Mouse | ↓ Accumbens DA | [125] |
| Rimonabant | 2, 10 mg/kg, i.p. | Rat | No effect on accumbens DA | [76] |
| Rimonabant | 1, 10, 30, 100 mM, intra-NAc | Rat | ↑ Accumbens DA | [76] |
| Rimonabant | 5, 10, 20 mg/kg, i.p. | Rat | ↑ Accumbens DA | [126] |
References
- Rudd, R.A.; Aleshire, N.; Zibbell, J.E.; Gladden, R.M. Increases in drug and opioid overdose deaths—United States, 2000–2014. MMWR Morb. Mortal Wkly. Rep. 2016, 64, 1378–1382.
- CDC (Center for Disease Control and Prevention). Drug over Dose Deaths; CDC: Atlanta, GA, USA, 2017.
- Jordan, C.J.; Xi, Z.X. Progress in brain cannabinoid CB2 receptor research: From genes to behavior. Neurosci. Biobehav. Rev. 2019, 98, 208–220.
- Jordan, C.J.; Xi, Z.X. Discovery and development of varenicline for smoking cessation. Expert Opin. Drug Discov. 2018, 13, 671–683.
- Galaj, E.; Newman, A.H.; Xi, Z.X. Dopamine D3 receptor-based medication development for the treatment of opioid use disorder: Rationale, progress, and challenges. Neurosci. Biobehav. Rev. 2020, 114, 38–52.
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773.
- Galaj, E.; Xi, Z.X. Potential of Cannabinoid Receptor Ligands as Treatment for Substance Use Disorders. CNS Drugs 2019, 33, 1001–1030.
- Le Foll, B.; Goldberg, S.R. Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence. J. Pharmacol. Exp. Ther. 2005, 312, 875–883.
- Sloan, M.E.; Gowin, J.L.; Ramchandani, V.A.; Hurd, Y.L.; Le Foll, B. The endocannabinoid system as a target for addiction treatment: Trials and tribulations. Neuropharmacology 2017, 124, 73–83.
- Butler, K.; Le Foll, B. Novel therapeutic and drug development strategies for tobacco use disorder: Endocannabinoid modulation. Expert. Opin. Drug Discov. 2020, 15, 1065–1080.
- Le Foll, B.; Gorelick, D.A.; Goldberg, S.R. The future of endocannabinoid-oriented clinical research after CB1 antagonists. Psychopharmacology 2009, 205, 171–174.
- Compton, W.M.; Wargo, E.M.; Volkow, N.D. Neuropsychiatric Model of Addiction Simplified. Psychiatr. Clin. N. Am. 2022, 45, 321–334.
- Galaj, E.; Hempel, B.; Moore, A.; Klein, B.; Bi, G.H.; Gardner, E.L.; Seltzman, H.H.; Xi, Z.X. Therapeutic potential of PIMSR, a novel CB1 receptor neutral antagonist, for cocaine use disorder: Evidence from preclinical research. Transl. Psychiatry 2022, 12, 286.
- Volkow, N.D.; Wise, R.A.; Baler, R. The dopamine motive system: Implications for drug and food addiction. Nat. Rev. Neurosci. 2017, 18, 741–752.
- Xi, Z.X.; Gardner, E.L. Hypothesis-driven medication discovery for the treatment of psychostimulant addiction. Curr. Drug Abuse Rev. 2008, 1, 303–327.
- Xi, Z.X.; Spiller, K.; Gardner, E.L. Mechanism-based medication development for the treatment of nicotine dependence. Acta Pharmacol. Sin. 2009, 30, 723–739.
- Jordan, C.J.; Cao, J.; Newman, A.H.; Xi, Z.X. Progress in agonist therapy for substance use disorders: Lessons learned from methadone and buprenorphine. Neuropharmacology 2019, 158, 107609.
- Wise, R.A. Brain reward circuitry: Insights from unsensed incentives. Neuron 2002, 36, 229–240.
- Ron, D.; Wang, J. The NMDA Receptor and Alcohol Addiction. In Biology of the NMDA Receptor; Van Dongen, A.M., Ed.; Frontiers in Neuroscience: Boca Raton, FL, USA, 2009; Chapter 4.
- Berrettini, W. Alcohol addiction and the mu-opioid receptor. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 65, 228–233.
- Di Chiara, G. Alcohol and dopamine. Alcohol Health Res. World 1997, 21, 108–114.
- Galaj, E.; Xi, Z.X. Progress in opioid reward research: From a canonical two-neuron hypothesis to two neural circuits. Pharmacol. Biochem. Behav. 2021, 200, 173072.
- Hempel, B.; Xi, Z.X. Receptor mechanisms underlying the CNS effects of cannabinoids: CB1 receptor and beyond. Adv. Pharmacol. 2022, 93, 275–333.
- Mackie, K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb. Exp. Pharmacol. 2005, 168, 299–325.
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949.
- McPartland, J.M.; Glass, M.; Pertwee, R.G. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: Interspecies differences. Br. J. Pharmacol. 2007, 152, 583–593.
- Hillard, C.J.; Edgemond, W.S.; Jarrahian, A.; Campbell, W.B. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J. Neurochem. 1997, 69, 631–638.
- Sugiura, T.; Waku, K. 2-Arachidonoylglycerol and the cannabinoid receptors. Chem. Phys. Lipids 2000, 108, 89–106.
- Trangenstein, P.J.; Whitehill, J.M.; Jenkins, M.C.; Jernigan, D.H.; Moreno, M.A. Cannabis Marketing and Problematic Cannabis Use Among Adolescents. J. Stud. Alcohol Drugs 2021, 82, 288–296.
- Fattore, L.; Fadda, P.; Spano, M.S.; Pistis, M.; Fratta, W. Neurobiological mechanisms of cannabinoid addiction. Mol. Cell Endocrinol. 2008, 286, S97–S107.
- Maldonado, R.; Valverde, O.; Berrendero, F. Involvement of the endocannabinoid system in drug addiction. Trends Neurosci. 2006, 29, 225–232.
- NSDUH. National Survey on Drug Use and Health. 2019. Available online: https://datafiles.samhsa.gov/ (accessed on 20 August 2022).
- D’Souza, D.C.; Perry, E.; MacDougall, L.; Ammerman, Y.; Cooper, T.; Wu, Y.T.; Braley, G.; Gueorguieva, R.; Krystal, J.H. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: Implications for psychosis. Neuropsychopharmacology 2004, 29, 1558–1572.
- Raft, D.; Gregg, J.; Ghia, J.; Harris, L. Effects of intravenous tetrahydrocannabinol on experimental and surgical pain. Psychological correlates of the analgesic response. Clin. Pharmacol. Ther. 1977, 21, 26–33.
- Lac, A.; Luk, J.W. Testing the Amotivational Syndrome: Marijuana Use Longitudinally Predicts Lower Self-Efficacy Even After Controlling for Demographics, Personality, and Alcohol and Cigarette Use. Prev. Sci. 2018, 19, 117–126.
- Petrucci, A.S.; LaFrance, E.M.; Cuttler, C. A Comprehensive Examination of the Links between Cannabis Use and Motivation. Subst. Use Misuse 2020, 55, 1155–1164.
- Barch, D.M.; Dowd, E.C. Goal representations and motivational drive in schizophrenia: The role of prefrontal-striatal interactions. Schizophr. Bull. 2010, 36, 919–934.
- Murray, C.H.; Glazer, J.E.; Lee, R.; Nusslock, R.; de Wit, H. Delta9-THC reduces reward-related brain activity in healthy adults. Psychopharmacology 2022, 239, 2829–2840.
- van Hell, H.H.; Jager, G.; Bossong, M.G.; Brouwer, A.; Jansma, J.M.; Zuurman, L.; van Gerven, J.; Kahn, R.S.; Ramsey, N.F. Involvement of the endocannabinoid system in reward processing in the human brain. Psychopharmacology 2012, 219, 981–990.
- Jansma, J.M.; van Hell, H.H.; Vanderschuren, L.J.; Bossong, M.G.; Jager, G.; Kahn, R.S.; Ramsey, N.F. THC reduces the anticipatory nucleus accumbens response to reward in subjects with a nicotine addiction. Transl. Psychiatry 2013, 3, e234.
- Lawn, W.; Freeman, T.P.; Pope, R.A.; Joye, A.; Harvey, L.; Hindocha, C.; Mokrysz, C.; Moss, A.; Wall, M.B.; Bloomfield, M.A.; et al. Acute and chronic effects of cannabinoids on effort-related decision-making and reward learning: An evaluation of the cannabis ‘amotivational’ hypotheses. Psychopharmacology 2016, 233, 3537–3552.
- Freeman, T.P.; Pope, R.A.; Wall, M.B.; Bisby, J.A.; Luijten, M.; Hindocha, C.; Mokrysz, C.; Lawn, W.; Moss, A.; Bloomfield, M.A.P.; et al. Cannabis Dampens the Effects of Music in Brain Regions Sensitive to Reward and Emotion. Int. J. Neuropsychopharmacol. 2018, 21, 21–32.
- Bloomfield, M.A.P.; Hindocha, C.; Green, S.F.; Wall, M.B.; Lees, R.; Petrilli, K.; Costello, H.; Ogunbiyi, M.O.; Bossong, M.G.; Freeman, T.P. The neuropsychopharmacology of cannabis: A review of human imaging studies. Pharmacol. Ther. 2019, 195, 132–161.
- Justinova, Z.; Tanda, G.; Redhi, G.H.; Goldberg, S.R. Self-administration of delta9-tetrahydrocannabinol (THC) by drug naive squirrel monkeys. Psychopharmacology 2003, 169, 135–140.
- Tanda, G.; Munzar, P.; Goldberg, S.R. Self-administration behavior is maintained by the psychoactive ingredient of marijuana in squirrel monkeys. Nat. Neurosci. 2000, 3, 1073–1074.
- Harris, R.T.; Waters, W.; McLendon, D. Evaluation of reinforcing capability of delta-9-tetrahydrocannabinol in rhesus monkeys. Psychopharmacologia 1974, 37, 23–29.
- John, W.S.; Martin, T.J.; Nader, M.A. Behavioral Determinants of Cannabinoid Self-Administration in Old World Monkeys. Neuropsychopharmacology 2017, 42, 1522–1530.
- Mansbach, R.S.; Nicholson, K.L.; Martin, B.R.; Balster, R.L. Failure of Delta(9)-tetrahydrocannabinol and CP 55,940 to maintain intravenous self-administration under a fixed-interval schedule in rhesus monkeys. Behav. Pharmacol. 1994, 5, 219–225.
- Panagis, G.; Vlachou, S.; Nomikos, G.G. Behavioral pharmacology of cannabinoids with a focus on preclinical models for studying reinforcing and dependence-producing properties. Curr. Drug Abuse Rev. 2008, 1, 350–374.
- Vlachou, S.; Panagis, G. Regulation of brain reward by the endocannabinoid system: A critical review of behavioral studies in animals. Curr. Pharm. Des. 2014, 20, 2072–2088.
- Spencer, S.; Neuhofer, D.; Chioma, V.C.; Garcia-Keller, C.; Schwartz, D.J.; Allen, N.; Scofield, M.D.; Ortiz-Ithier, T.; Kalivas, P.W. A Model of Delta(9)-Tetrahydrocannabinol Self-administration and Reinstatement That Alters Synaptic Plasticity in Nucleus Accumbens. Biol. Psychiatry 2018, 84, 601–610.
- Neuhofer, D.; Spencer, S.M.; Chioma, V.C.; Beloate, L.N.; Schwartz, D.; Kalivas, P.W. The loss of NMDAR-dependent LTD following cannabinoid self-administration is restored by positive allosteric modulation of CB1 receptors. Addict. Biol. 2020, 25, e12843.
- Cheer, J.F.; Kendall, D.A.; Marsden, C.A. Cannabinoid receptors and reward in the rat: A conditioned place preference study. Psychopharmacology 2000, 151, 25–30.
- DeVuono, M.V.; Wills, K.L.; MacPherson, D.V.; Hrelja, K.M.; Parker, L.A. Effect of footshock stress on place conditioning produced by Delta(9)-tetrahydrocannabinol and the fatty acid amide hydrolase (FAAH) inhibitor, URB597, in Sprague-Dawley rats. Psychopharmacology 2017, 234, 3229–3240.
- Braida, D.; Iosue, S.; Pegorini, S.; Sala, M. Delta9-tetrahydrocannabinol-induced conditioned place preference and intracerebroventricular self-administration in rats. Eur. J. Pharmacol. 2004, 506, 63–69.
- Lepore, M.; Liu, X.; Savage, V.; Matalon, D.; Gardner, E.L. Genetic differences in delta 9-tetrahydrocannabinol-induced facilitation of brain stimulation reward as measured by a rate-frequency curve-shift electrical brain stimulation paradigm in three different rat strains. Life Sci. 1996, 58, PL365–PL372.
- Gardner, E.L.; Paredes, W.; Smith, D.; Donner, A.; Milling, C.; Cohen, D.; Morrison, D. Facilitation of brain stimulation reward by delta 9-tetrahydrocannabinol. Psychopharmacology 1988, 96, 142–144.
- Katsidoni, V.; Kastellakis, A.; Panagis, G. Biphasic effects of Delta9-tetrahydrocannabinol on brain stimulation reward and motor activity. Int. J. Neuropsychopharmacol. 2013, 16, 2273–2284.
- Kwilasz, A.J.; Negus, S.S. Dissociable effects of the cannabinoid receptor agonists Delta9-tetrahydrocannabinol and CP55940 on pain-stimulated versus pain-depressed behavior in rats. J. Pharmacol. Exp. Ther. 2012, 343, 389–400.
- Negus, S.S.; Miller, L.L. Intracranial self-stimulation to evaluate abuse potential of drugs. Pharmacol. Rev. 2014, 66, 869–917.
- Vlachou, S.; Nomikos, G.G.; Stephens, D.N.; Panagis, G. Lack of evidence for appetitive effects of Delta 9-tetrahydrocannabinol in the intracranial self-stimulation and conditioned place preference procedures in rodents. Behav. Pharmacol. 2007, 18, 311–319.
- Wiebelhaus, J.M.; Grim, T.W.; Owens, R.A.; Lazenka, M.F.; Sim-Selley, L.J.; Abdullah, R.A.; Niphakis, M.J.; Vann, R.E.; Cravatt, B.F.; Wiley, J.L.; et al. Delta9-tetrahydrocannabinol and endocannabinoid degradative enzyme inhibitors attenuate intracranial self-stimulation in mice. J. Pharmacol. Exp. Ther. 2015, 352, 195–207.
- Spiller, K.J.; Bi, G.H.; He, Y.; Galaj, E.; Gardner, E.L.; Xi, Z.X. Cannabinoid CB1 and CB2 receptor mechanisms underlie cannabis reward and aversion in rats. Br. J. Pharmacol. 2019, 176, 1268–1281.
- Han, X.; He, Y.; Bi, G.H.; Zhang, H.Y.; Song, R.; Liu, Q.R.; Egan, J.M.; Gardner, E.L.; Li, J.; Xi, Z.X. CB1 Receptor Activation on VgluT2-Expressing Glutamatergic Neurons Underlies Delta(9)-Tetrahydrocannabinol (Delta(9)-THC)-Induced Aversive Effects in Mice. Sci. Rep. 2017, 7, 12315.
- Humburg, B.A.; Jordan, C.J.; Zhang, H.Y.; Shen, H.; Han, X.; Bi, G.H.; Hempel, B.; Galaj, E.; Baumann, M.H.; Xi, Z.X. Optogenetic brain-stimulation reward: A new procedure to re-evaluate the rewarding versus aversive effects of cannabinoids in dopamine transporter-Cre mice. Addict. Biol. 2021, 26, e13005.
- Cheer, J.F.; Kendall, D.A.; Mason, R.; Marsden, C.A. Differential cannabinoid-induced electrophysiological effects in rat ventral tegmentum. Neuropharmacology 2003, 44, 633–641.
- Cheer, J.F.; Wassum, K.M.; Heien, M.L.; Phillips, P.E.; Wightman, R.M. Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J. Neurosci. 2004, 24, 4393–4400.
- Tanda, G.; Pontieri, F.E.; Di Chiara, G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science 1997, 276, 2048–2050.
- Covey, D.P.; Mateo, Y.; Sulzer, D.; Cheer, J.F.; Lovinger, D.M. Endocannabinoid modulation of dopamine neurotransmission. Neuropharmacology 2017, 124, 52–61.
- Fitzgerald, M.L.; Shobin, E.; Pickel, V.M. Cannabinoid modulation of the dopaminergic circuitry: Implications for limbic and striatal output. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 38, 21–29.
- Paladini, C.A.T. Neurophysiology of substantia nigra dopamine neurons: Modulation by GABA and glutamate. In Handbook of Basal Ganglia Structure and Function; Elsevier: Amsterdam, The Netherlands, 2017; pp. 335–360.
- Szabo, B.; Muller, T.; Koch, H. Effects of cannabinoids on dopamine release in the corpus striatum and the nucleus accumbens in vitro. J. Neurochem. 1999, 73, 1084–1089.
- Castañeda, E.M.; Oddie, S.D.; Whishaw, Q. THC does not affect striatal dopamine release: Microdialysis in freely moving rats. Pharmacol. Biochem. Behav. 1991, 40, 587–591.
- Pillolla, G.; Melis, M.; Perra, S.; Muntoni, A.L.; Gessa, G.L.; Pistis, M. Medial forebrain bundle stimulation evokes endocannabinoid-mediated modulation of ventral tegmental area dopamine neuron firing in vivo. Psychopharmacology 2007, 191, 843–853.
- Sidlo, Z.; Reggio, P.H.; Rice, M.E. Inhibition of striatal dopamine release by CB1 receptor activation requires nonsynaptic communication involving GABA, H2O2, and KATP channels. Neurochem. Int. 2008, 52, 80–88.
- Li, X.; Hempel, B.J.; Yang, H.J.; Han, X.; Bi, G.H.; Gardner, E.L.; Xi, Z.X. Dissecting the role of CB1 and CB2 receptors in cannabinoid reward versus aversion using transgenic CB1- and CB2-knockout mice. Eur. Neuropsychopharmacol. 2021, 43, 38–51.
- Lupica, C.R.; Riegel, A.C. Endocannabinoid release from midbrain dopamine neurons: A potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology 2005, 48, 1105–1116.
- Szabo, B.; Siemes, S.; Wallmichrath, I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur. J. Neurosci. 2002, 15, 2057–2061.
- Melis, M.; Pistis, M. Endocannabinoid signaling in midbrain dopamine neurons: More than physiology? Curr. Neuropharmacol. 2007, 5, 268–277.
- Melis, M.; Sagheddu, C.; De Felice, M.; Casti, A.; Madeddu, C.; Spiga, S.; Muntoni, A.L.; Mackie, K.; Marsicano, G.; Colombo, G.; et al. Enhanced endocannabinoid-mediated modulation of rostromedial tegmental nucleus drive onto dopamine neurons in Sardinian alcohol-preferring rats. J. Neurosci. 2014, 34, 12716–12724.
- Wang, X.F.; Galaj, E.; Bi, G.H.; Zhang, C.; He, Y.; Zhan, J.; Bauman, M.H.; Gardner, E.L.; Xi, Z.X. Different receptor mechanisms underlying phytocannabinoid- versus synthetic cannabinoid-induced tetrad effects: Opposite roles of CB1 /CB2 versus GPR55 receptors. Br. J. Pharmacol. 2020, 177, 1865–1880.
- Manzanares, J.; Cabanero, D.; Puente, N.; Garcia-Gutierrez, M.S.; Grandes, P.; Maldonado, R. Role of the endocannabinoid system in drug addiction. Biochem. Pharmacol. 2018, 157, 108–121.
- Zhang, H.Y.; Gao, M.; Liu, Q.R.; Bi, G.H.; Li, X.; Yang, H.J.; Gardner, E.L.; Wu, J.; Xi, Z.X. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E5007–E5015.
- Zhang, H.Y.; Gao, M.; Shen, H.; Bi, G.H.; Yang, H.J.; Liu, Q.R.; Wu, J.; Gardner, E.L.; Bonci, A.; Xi, Z.X. Expression of functional cannabinoid CB2 receptor in VTA dopamine neurons in rats. Addict. Biol. 2017, 22, 752–765.
- Xi, Z.X.; Peng, X.Q.; Li, X.; Song, R.; Zhang, H.Y.; Liu, Q.R.; Yang, H.J.; Bi, G.H.; Li, J.; Gardner, E.L. Brain cannabinoid CB(2) receptors modulate cocaine’s actions in mice. Nat. Neurosci. 2011, 14, 1160–1166.
- Foster, D.J.; Wilson, J.M.; Remke, D.H.; Mahmood, M.S.; Uddin, M.J.; Wess, J.; Patel, S.; Marnett, L.J.; Niswender, C.M.; Jones, C.K.; et al. Antipsychotic-like Effects of M4 Positive Allosteric Modulators Are Mediated by CB2 Receptor-Dependent Inhibition of Dopamine Release. Neuron 2016, 91, 1244–1252.
- Zhang, H.Y.; Bi, G.H.; Li, X.; Li, J.; Qu, H.; Zhang, S.J.; Li, C.Y.; Onaivi, E.S.; Gardner, E.L.; Xi, Z.X.; et al. Species differences in cannabinoid receptor 2 and receptor responses to cocaine self-administration in mice and rats. Neuropsychopharmacology 2015, 40, 1037–1051.
- Aracil-Fernandez, A.; Trigo, J.M.; Garcia-Gutierrez, M.S.; Ortega-Alvaro, A.; Ternianov, A.; Navarro, D.; Robledo, P.; Berbel, P.; Maldonado, R.; Manzanares, J. Decreased cocaine motor sensitization and self-administration in mice overexpressing cannabinoid CB(2) receptors. Neuropsychopharmacology 2012, 37, 1749–1763.
- Delis, F.; Polissidis, A.; Poulia, N.; Justinova, Z.; Nomikos, G.G.; Goldberg, S.R.; Antoniou, K. Attenuation of Cocaine-Induced Conditioned Place Preference and Motor Activity via Cannabinoid CB2 Receptor Agonism and CB1 Receptor Antagonism in Rats. Int. J. Neuropsychopharmacol. 2017, 20, 269–278.
- Cahill, K.; Ussher, M.H. Cannabinoid type 1 receptor antagonists for smoking cessation. Cochrane Database Syst. Rev. 2011, CD005353.
- Elrashidi, M.Y.; Ebbert, J.O. Emerging drugs for the treatment of tobacco dependence: 2014 update. Expert Opin. Emerg. Drugs 2014, 19, 243–260.
- Steinberg, M.B.; Foulds, J. Rimonabant for treating tobacco dependence. Vasc. Health Risk Manag. 2007, 3, 307–311.
- Huestis, M.A.; Boyd, S.J.; Heishman, S.J.; Preston, K.L.; Bonnet, D.; Le Fur, G.; Gorelick, D.A. Single and multiple doses of rimonabant antagonize acute effects of smoked cannabis in male cannabis users. Psychopharmacology 2007, 194, 505–515.
- Solinas, M.; Panlilio, L.V.; Goldberg, S.R. Exposure to delta-9-tetrahydrocannabinol (THC) increases subsequent heroin taking but not heroin’s reinforcing efficacy: A self-administration study in rats. Neuropsychopharmacology 2004, 29, 1301–1311.
- Gamaleddin, I.; Wertheim, C.; Zhu, A.Z.; Coen, K.M.; Vemuri, K.; Makryannis, A.; Goldberg, S.R.; Le Foll, B. Cannabinoid receptor stimulation increases motivation for nicotine and nicotine seeking. Addict. Biol. 2012, 17, 47–61.
- Colombo, G.; Serra, S.; Brunetti, G.; Gomez, R.; Melis, S.; Vacca, G.; Carai, M.M.; Gessa, L. Stimulation of voluntary ethanol intake by cannabinoid receptor agonists in ethanol-preferring sP rats. Psychopharmacology 2002, 159, 181–187.
- Linsenbardt, D.N.; Boehm, S.L., 2nd. Agonism of the endocannabinoid system modulates binge-like alcohol intake in male C57BL/6J mice: Involvement of the posterior ventral tegmental area. Neuroscience 2009, 164, 424–434.
- Gamaleddin, I.H.; Trigo, J.M.; Gueye, A.B.; Zvonok, A.; Makriyannis, A.; Goldberg, S.R.; Le Foll, B. Role of the endogenous cannabinoid system in nicotine addiction: Novel insights. Front. Psychiatry 2015, 6, 41.
- Maccioni, P.; Colombo, G.; Carai, M.A. Blockade of the cannabinoid CB1 receptor and alcohol dependence: Preclinical evidence and preliminary clinical data. CNS Neurol. Disord. Drug Targets 2010, 9, 55–59.
- Solinas, M.; Panlilio, L.V.; Antoniou, K.; Pappas, L.A.; Goldberg, S.R. The cannabinoid CB1 antagonist N-piperidinyl-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl) -4-methylpyrazole-3-carboxamide (SR-141716A) differentially alters the reinforcing effects of heroin under continuous reinforcement, fixed ratio, and progressive ratio schedules of drug self-administration in rats. J. Pharmacol. Exp. Ther. 2003, 306, 93–102.
- De Vries, T.J.; Homberg, J.R.; Binnekade, R.; Raaso, H.; Schoffelmeer, A.N.M. Cannabinoid modulation of the reinforcing and motivational properties of heroin and heroin-associated cues in rats. Psychopharmacology 2003, 168, 164–169.
- Cohen, C.; Perrault, G.; Voltz, C.; Steinberg, R.; Soubrie, P. SR141716, a central cannabinoid (CB(1)) receptor antagonist, blocks the motivational and dopamine-releasing effects of nicotine in rats. Behav. Pharmacol. 2002, 13, 451–463.
- Cohen, C.; Perrault, G.; Griebel, G.; Soubrie, P. Nicotine-associated cues maintain nicotine-seeking behavior in rats several weeks after nicotine withdrawal: Reversal by the cannabinoid (CB1) receptor antagonist, rimonabant (SR141716). Neuropsychopharmacology 2005, 30, 145–155.
- Le Foll, B.; Goldberg, S.R. Rimonabant, a CB1 antagonist, blocks nicotine-conditioned place preferences. Neuroreport. 2004, 15, 2139–2143.
- Chaperon, F.; Soubrie, P.; Puech, A.J.; Thiebot, M.H. Involvement of central cannabinoid (CB1) receptors in the establishment of place conditioning in rats. Psychopharmacology 1998, 135, 324–332.
- De Vries, T.J.; Shaham, Y.; Homberg, J.R.; Crombag, H.; Schuurman, K.; Dieben, J.; Vanderschuren, L.J.; Schoffelmeer, A.N. A cannabinoid mechanism in relapse to cocaine seeking. Nat. Med. 2001, 7, 1151–1154.
- Anggadiredja, K.; Nakamichi, M.; Hiranita, T.; Tanaka, H.; Shoyama, Y.; Watanabe, S.; Yamamoto, T. Endocannabinoid system modulates relapse to methamphetamine seeking: Possible mediation by the arachidonic acid cascade. Neuropsychopharmacology 2004, 29, 1470–1478.
- Henderson-Redmond, A.N.; Guindon, J.; Morgan, D.J. Roles for the endocannabinoid system in ethanol-motivated behavior. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 65, 330–339.
- Nguyen, T.; Thomas, B.F.; Zhang, Y. Overcoming the Psychiatric Side Effects of the Cannabinoid CB1 Receptor Antagonists: Current Approaches for Therapeutics Development. Curr. Top. Med. Chem. 2019, 19, 1418–1435.
- Gonzalez, S.; Schmid, P.C.; Fernandez-Ruiz, J.; Krebsbach, R.; Schmid, H.H.; Ramos, J.A. Region-dependent changes in endocannabinoid transmission in the brain of morphine-dependent rats. Addict. Biol. 2003, 8, 159–166.
- Vigano, D.; Grazia Cascio, M.; Rubino, T.; Fezza, F.; Vaccani, A.; Di Marzo, V.; Parolaro, D. Chronic morphine modulates the contents of the endocannabinoid, 2-arachidonoyl glycerol, in rat brain. Neuropsychopharmacology 2003, 28, 1160–1167.
- Vigano, D.; Valenti, M.; Cascio, M.G.; Di Marzo, V.; Parolaro, D.; Rubino, T. Changes in endocannabinoid levels in a rat model of behavioural sensitization to morphine. Eur. J. Neurosci. 2004, 20, 1849–1857.
- Gonzalez, S.; Cascio, M.G.; Fernandez-Ruiz, J.; Fezza, F.; Di Marzo, V.; Ramos, J.A. Changes in endocannabinoid contents in the brain of rats chronically exposed to nicotine, ethanol or cocaine. Brain Res. 2002, 954, 73–81.
- Wang, H.; Treadway, T.; Covey, D.P.; Cheer, J.F.; Lupica, C.R. Cocaine-Induced Endocannabinoid Mobilization in the Ventral Tegmental Area. Cell Rep. 2015, 12, 1997–2008.
- Caille, S.; Alvarez-Jaimes, L.; Polis, I.; Stouffer, D.G.; Parsons, L.H. Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J. Neurosci. 2007, 27, 3695–3702.
- Zhang, H.; Lipinski, A.A.; Liktor-Busa, E.; Smith, A.F.; Moutal, A.; Khanna, R.; Langlais, P.R.; Largent-Milnes, T.M.; Vanderah, T.W. The Effects of Repeated Morphine Treatment on the Endogenous Cannabinoid System in the Ventral Tegmental Area. Front. Pharmacol. 2021, 12, 632757.
- Buczynski, M.W.; Polis, I.Y.; Parsons, L.H. The volitional nature of nicotine exposure alters anandamide and oleoylethanolamide levels in the ventral tegmental area. Neuropsychopharmacology 2013, 38, 574–584.
- He, X.H.; Jordan, C.J.; Vemuri, K.; Bi, G.H.; Zhan, J.; Gardner, E.L.; Makriyannis, A.; Wang, Y.L.; Xi, Z.X. Cannabinoid CB1 receptor neutral antagonist AM4113 inhibits heroin self-administration without depressive side effects in rats. Acta Pharmacol. Sin. 2019, 40, 365–373.
- Anisman, H.; Matheson, K. Stress, depression, and anhedonia: Caveats concerning animal models. Neurosci. Biobehav. Rev. 2005, 29, 525–546.
- Scheggi, S.; De Montis, M.G.; Gambarana, C. Making Sense of Rodent Models of Anhedonia. Int. J. Neuropsychopharmacol. 2018, 21, 1049–1065.
- Arnold, J.C.; Hunt, G.E.; McGregor, I.S. Effects of the cannabinoid receptor agonist CP 55,940 and the cannabinoid receptor antagonist SR 141716 on intracranial self-stimulation in Lewis rats. Life Sci. 2001, 70, 97–108.
- Grim, T.W.; Wiebelhaus, J.M.; Morales, A.J.; Negus, S.S.; Lichtman, A.H. Effects of acute and repeated dosing of the synthetic cannabinoid CP55,940 on intracranial self-stimulation in mice. Drug Alcohol Depend. 2015, 150, 31–37.
- Vlachou, S.; Nomikos, G.G.; Panagis, G. Effects of endocannabinoid neurotransmission modulators on brain stimulation reward. Psychopharmacology 2006, 188, 293–305.
- Vlachou, S.; Stamatopoulou, F.; Nomikos, G.G.; Panagis, G. Enhancement of endocannabinoid neurotransmission through CB1 cannabinoid receptors counteracts the reinforcing and psychostimulant effects of cocaine. Int. J. Neuropsychopharmacol. 2008, 11, 905–923.
- Li, X.; Hoffman, A.F.; Peng, X.Q.; Lupica, C.R.; Gardner, E.L.; Xi, Z.X. Attenuation of basal and cocaine-enhanced locomotion and nucleus accumbens dopamine in cannabinoid CB1-receptor-knockout mice. Psychopharmacology 2009, 204, 1–11.
- Murillo-Rodriguez, E.; Machado, S.; Rocha, N.B.; Budde, H.; Yuan, T.F.; Arias-Carrion, O. Revealing the role of the endocannabinoid system modulators, SR141716A, URB597 and VDM-11, in sleep homeostasis. Neuroscience 2016, 339, 433–449.
- Li, F.; Fang, Q.; Liu, Y.; Zhao, M.; Li, D.; Wang, J.; Lu, L. Cannabinoid CB(1) receptor antagonist rimonabant attenuates reinstatement of ketamine conditioned place preference in rats. Eur. J. Pharmacol. 2008, 589, 122–126.
- Biala, G.; Budzynska, B.; Staniak, N. Effects of rimonabant on the reinstatement of nicotine-conditioned place preference by drug priming in rats. Behav. Brain. Res. 2009, 202, 260–265.
- Singh, M.E.; Verty, A.N.; McGregor, I.S.; Mallet, P.E. A cannabinoid receptor antagonist attenuates conditioned place preference but not behavioural sensitization to morphine. Brain Res. 2004, 1026, 244–253.
- Fang, Q.; Li, F.Q.; Li, Y.Q.; Xue, Y.X.; He, Y.Y.; Liu, J.F.; Lu, L.; Wang, J.S. Cannabinoid CB1 receptor antagonist rimonabant disrupts nicotine reward-associated memory in rats. Pharmacol. Biochem. Behav. 2011, 99, 738–742.
More
Information
Subjects:
Biology; Physiology; Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Revisions:
2 times
(View History)
Update Date:
20 Jan 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No