Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Omar Soler-Cedeño and Version 2 by Rita Xu.
Cannabinoid receptor 1 (CB1R) has been one of the major targets in medication development for treating substance use disorders (SUDs). Early studies indicated that rimonabant, a selective CB1R antagonist with an inverse agonist profile, was highly promising as a therapeutic for SUDs.
- cannabinoid
- CB1 receptor
- Δ9-tetrahydrocannabinol
- rimonabant
- substance use disorders
1. Introduction
Substance use disorder (SUD), defined as the uncontrollable and persistent use of drugs (including alcohol) despite substantial harm and adverse consequences, is still a severe social and health problem worldwide. SUD-related costs, including those in crimes, loss of productivity and healthcare, exceed $740 billion per year in the Unites States [1]. In recent years, opioid overdose and SUD-related diseases have increased dramatically with the fatal incidents up to ~50,000 in 2017 in the USA [2]. Although the United States Food and Drug Administration (FDA) approved several medications such as methadone, buprenorphine, and varenicline for the treatment of opioid or nicotine use disorders [3][4][5][3,4,5], the rate of relapse remains extremely high. Moreover, there is no FDA-approved medication for the treatment of psychostimulant use disorders [6]. Over the past decades, the cannabinoid receptor 1 (CB1R) has been given much attention as a promising target in medication development for treating SUDs [7][8][9][7,8,9]. The reason for such attention is because of convincing evidence indicating that rimonabant, a selective CB1R antagonist with an inverse agonist profile, is highly effective in reducing drug taking and drug-seeking behavior in experimental animals [7][8][10][7,8,10]. However, the severe adverse effects of rimonabant, such as nausea, emesis, depression, and suicidal tendencies observed in humans have led to its withdrawal from clinical trials worldwide [11]. Consequently, the US FDA decided not to approve CB1R ligands until better safety and efficacy data become available.
2. Mesocorticolimbic Dopamine System
To better understand how cannabinoid CB1R antagonists produce anti-addictive effects, it is necessary to briefly review the current working hypothesis underlying drug reward and addiction. Addiction includes three stages—binge/intoxication, a stage at which an individual consumes an intoxicating substance and experiences its rewarding effects; withdrawal/negative affect, a stage at which an individual experiences a negative emotional state in the absence of the substance; and preoccupation/anticipation, a stage at which subject seeks substances again after a period of abstinence [12]. Although theour understanding of the neural mechanisms underlying each stage of addiction is still not fully understood, a well-accepted view is that the rewarding effects of drugs of abuse are mediated mainly by activation of the mesocorticolimbic dopamine (DA) system. This system originates in DA neurons in the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc) of the midbrain and projects to the prefrontal cortex (PFC), nucleus accumbens (NAc), and the dorsal striatum (SD) [6][13][6,13]. Different drugs of abuse activate this pathway by distinct receptor and cellular mechanisms [14][15][16][14,15,16] (Figure 1). For example, the psychostimulant cocaine activates this system mainly by blocking the DA transporter (DAT), while nicotine activates VTA DA neurons by stimulating nicotinic receptors located on DA neurons or glutamate neurons that project to DA neurons in the VTA and NAc [16][17][18][16,17,18]. Alcohol’s reinforcement has been associated with processes involving multiple molecular targets, including mu opioid receptors and NMDA receptors [12][19][20][21][12,19,20,21]. On the other hand, opioids activate midbrain DA neurons mainly by stimulation of opioid receptors located on GABAergic neurons in the rostromedial tegmentum (RMTg) and substantia nigra pars reticulata (SNr) that project to the VTA and SNc, respectively, causing increases in DA neurons firing and striatal DA release via GABA-mediated disinhibition [5][17][22][5,17,22]. Therefore, both the RMTg-VTA-NAc and SNr-SNc-DS DA pathways play a central role in drug reward and addiction [14][22][14,22], making the DA system a crucial target in medication development for the treatment of SUDs.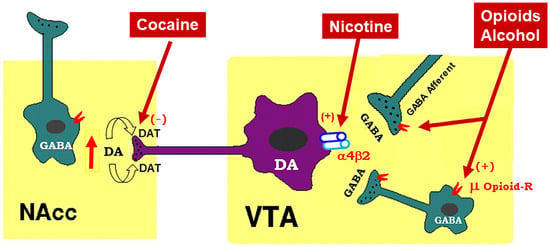
Figure 1. Schematic diagram of the mesolimbic dopamine (DA) hypothesis, illustrating how drugs of abuse activate this system. The mesolimbic DA system originates in the midbrain ventral tegmental area (VTA) and projects predominantly to the forebrain nucleus accumbens (NAc) and the prefrontal cortex (not shown). The psychostimulant cocaine elevates extracellular NAc DA by blocking DA transporters (DAT) on DA axon terminals, while opioids (such as heroin) and alcohol bind to and activate mu opioid receptor (μ Opioid–R) located mainly on GABAergic afferents (less on VTA GABAergic interneurons) and inhibit GABA release. A reduction in GABA release leads to DA neuron disinhibition (activation). Nicotine has been thought to activate DA neurons mainly by activation of α4β2 nicotinic receptors located on DA neurons. (+), (-): indicate activation of opiate receptors or blockade of DAT.
3. Endocannabinoid System
To better understand how CB1R antagonists produce therapeutic effects against drug abuse and addiction and how CB1R inverse agonists produce unwanted side-effects, let reusearchers briefly review the endocannabinoid (eCB) system and recent research on how cannabinoids modulate the mesocorticolimbic DA system, a critical action site for drugs of abuse. The endocannabinoid (eCB) system consists of cannabinoid receptors (CB1Rs, CB2Rs, and others), endocannabinoids [anandamide (AEA) and 2-arachidonoylglycerol (2-AG)], enzymes for endocannabinoid synthesis [N-arachidonoyl phosphatidylethanolamine-phospholipase D (NAPE-PLD), diacylglycerol-lipase (DAG-lipase)] and degradation [fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MGL)], and their putative transport systems [23][24][23,24]. AEA was the first endocannabinoid discovered by Raphael Mechoulam and his colleagues in 1992 [25]. AEA is an endogenous CB1R agonist (Ki = 87.7 nM for rCB1; Ki = 239.2 nM for hCB1) and a weak CB2R agonist (Ki = 267.8 nM for rCB2; Ki = 439.5 nM for hCB2) [26]. The effects of AEA are mediated mainly by activation of CB1Rs and CB2Rs in the brain and periphery. However, AEA levels in the brain are very low in healthy subjects, and it has a very short half-life (~2 min) due to its fast degradation by fatty acid amide hydroxylase (FAAH) [27]. Therefore, the functional significance of AEA in the brain is largely unclear. 2-AG was the second eCB discovered in the brain [28]. It is an endogenous agonist of the CB1Rs (Ki = 1180 nM for rCB1; Ki = 3423 nM for hCB1) and CB2Rs (Ki = 1900 nM for rCB2; Ki = 1193 nM for hCB2) [26]. Unlike AEA, 2-AG is present at relatively high levels in the central nervous system (CNS). Therefore, it is thought to be a major eCB modulating brain function. There are at least two types of cannabinoid receptors (CB1Rs and CB2Rs) identified in the brain [23]. The phytocannabinoids (Δ9-THC), synthetic cannabinoids (WIN55,212-2, CP55,940, HU-210), and the endocannabinoids (AEA, 2-AG) all have high binding affinities at both the CB1Rs and CB2Rs [23]. Cannabinoids may also bind to other putative cannabinoid receptors, such as G protein-coupled receptor 55 (GPR55), transient receptor potential vanilloid 1 (TRPV1) channel, and peroxisome proliferator-activated nuclear receptors (PPARs) [23]. Accumulative evidence indicates that cannabinoid action is mediated mainly by activation of CB1Rs and CB2Rs [23].4. Cannabinoid Reward versus Aversion
Cannabis is the most commonly used substance worldwide as many people find it pleasurable [29]. However, the findings regarding the rewarding properties of cannabinoids in both humans and experimental animals are conflicting [23]. Indeed, cannabis use has often been associated with its psychoactive, rewarding effects [30][31][30,31]. The psychoactive effects of cannabis, combined with the ongoing cannabis legalization in the United States, may well explain why cannabis use is rising in the USA. For instance, from 2002 to 2019, the percentage of adults who reported using cannabis in the past year increased from 7.0 to 15.2% [32]. However, cannabis enjoyment is not universal, and some individuals report dysphoria, anxiety, and depression after cannabis use [33][34][33,34]. The increase in cannabis use also raises concerns about possible adverse effects of cannabis use, such as developing the amotivational syndrome [35][36][35,36], which is defined as “a reduction in the motivation to initiate or persist in goal-directed behavior” [37]. A series of human functional magnetic resonance imaging (fMRI) studies support these cannabis amotivational effects by evidence that Δ9-THC produces a significant reduction in reward-related brain activity or neural response to reward in healthy adults [38][39][40][38,39,40]. In congruent with these findings, other reports showed that Δ9-THC reduced the likelihood or motivation of reward-related learning and decision-making [41], dampened neural responses to music [42], and reduced striatal DA response to reward [43]. Similar paradoxical effects of cannabinoids have been discovered in non-human primates, as squirrel monkeys self-administer Δ9-THC or endocannabinoids [44][45][44,45], while other primate species (rhesus, baboon, cynomolgus) fail to demonstrate this behavior [46][47][48][46,47,48]. In rodents, Δ9-THC alone is not self-administered [49][50][49,50], although the mixture of Δ9-THC and cannabidiol was recently reported to be self-administered by rats [51][52][51,52]. In conditioned place preference (CPP) test, Δ9-THC typically produces conditioned place aversion [53][54][53,54], although place preferences have also been reported [55][56][55,56]. In electrical intracranial self-stimulation (ICSS) experiments, Δ9-THC was initially reported to facilitate electrical ICSS in rats [56][57][58][56,57,58], while other studies found suppression of ICSS in rats and mice [59][60][61][62][63][59,60,61,62,63]. In optogenetic ICSS (oICSS) maintained by optical stimulation of midbrain DA neurons or glutamate neurons, cannabinoids always produce a reduction in brain-stimulation reward (BSR) in mice, suggesting a reward-attenuating or aversive effect [64][65][64,65]. Similarly, the findings of cannabinoid action on DA transmission are also conflicting. There are reports indicating that activation of the CB1Rs increases DA neuronal firing in the VTA [66][67][66,67] and DA release in the NAc in rats [68][69][70][71][68,69,70,71]. However, in vitro voltammetry experiments in striatal brain slices demonstrate that the cannabinoids WIN55,212-2 or CP55,940 fail to alter [72][73][72,73] or produce a reduction in electrical stimulation-induced DA release in the dorsal striatum in guinea pigs, rats and mice [74][75][74,75]. In vivo microdialysis experiments in freely moving animals indicate that Δ9-THC produces a dose-dependent reduction in NAc DA in mice [76]. The neural mechanisms underlying such opposite affective and neurochemical effects of cannabinoids are not fully understood.4.1. GABAergic CB1R Hypothesis of Cannabis Reward
Given that midbrain DA neurons receive both inhibitory GABAergic and excitatory glutamatergic inputs, reswearchers proposed that differential CB1R expression on GABAergic neurons versus glutamatergic neurons may underlie cannabinoid reward versus aversion, respectively [7][23][64][7,23,64] (Figure 2). The GABAergic CB1R hypothesis is supported by electrophysiological findings in brain slices where stimulation of CB1Rs on VTA GABAergic neurons causes an increase in VTA DA neuron firing via GABA-mediated disinhibition [77][78][79][80][77,78,79,80]. However, so far, there is a lack of behavioral evidence in vivo supporting this GABA-CB1R hypothesis possibly due to the absence of reliable behavioral models of cannabinoid reward in rodents.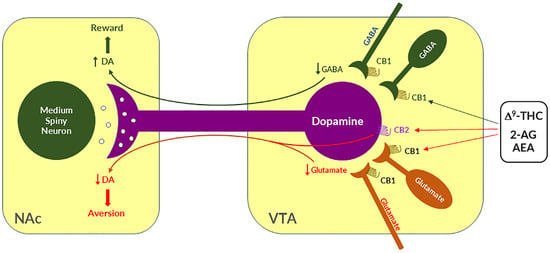
Figure 2. A schematic diagram showing the neural mechanisms underlying cannabis reward versus aversion. Cannabinoid CB1Rs are expressed not only in VTA GABAergic interneurons or GABAergic afferents but also in VTA glutamatergic neurons or their afferents within the VTA, whereas CB2Rs are also expressed in VTA DA neurons. Both CB1Rs and CB2Rs are inhibitory G-protein (Gi)-coupled receptors, producing an inhibitory effect on neuronal firing or terminal neurotransmitter release after activation. Cannabinoids such as Δ9-THC, 2-AG, and AEA may produce rewarding effects by binding to CB1Rs on VTA GABAergic interneurons and/or their afferents as a reduction in GABA release causes an increase DA neuronal firing and enhanced DA release in the NAc. Conversely, cannabinoids may also produce aversive effects by activating CB1Rs on glutamatergic neurons and/or terminals in the VTA that decreases excitatory glutamate input on VTA DA neurons. In addition, activation of CB2Rs on VTA DA neurons also produce an inhibitory effect on DA neuron firing and DA release in the NAc. Thus, the subjective effects of cannabinoids may depend on the balance of both oppose actions. This hypothesis may well explain why cannabinoids are rewarding in some subjects or species, while ineffective or even aversive in others. ↑, ↓—indicate an increase or a decrease in neurotransmitter release.
4.2. Glutamatergic CB1 Hypothesis of Cannabinoid Aversion
Clearly, the above GABAergic disinhibition hypothesis cannot explain how cannabinoids produce the aversive effects observed in rodents. To address this question, rwesearchers have recently used advanced RNAscope in situ hybridization (ISH) assays to examine the cellular distributions of CB1Rs. ThWey found that CB1Rs are expressed not only in VTA GABAergic neurons but also in VTA glutamatergic neurons [23][64][65][23,64,65]. Strikingly, optogenetic activation of VTA glutamatergic neurons produced potent rewarding effects, as assessed by CPP and optical ICSS (oICSS) [64][81][64,81]. Systemic administration of multiple cannabinoids (such as Δ9-THC, WIN55,212-2, ACEA, AM-2201) dose-dependently inhibited glutamate-mediated oICSS only in VgluT2-Cre control mice, but not in glutamate-CB1-knockout mice in which CB1Rs are selectively deleted from subcortical VgluT2-expressing glutamate neurons [64]. These findings suggest that activation of CB1Rs on glutamate neurons produces reward-attenuation or aversive effects by decreasing glutamatergic inputs onto VTA DA neurons (Figure 2). The above findings also suggest that activation of brain CB1Rs is not always rewarding but it could be aversive, depending upon the cellular distribution of CB1R expression in the brain. Therefore, reswearchers propose that the hedonic effects of cannabis might depend on the balance of two opposing actions of cannabinoids on both GABAergic neurons and glutamatergic neurons (Figure 2). If more CB1Rs are expressed in VTA or VTA-projecting GABAergic neurons, cannabis will be rewarding as GABAergic disinhibition of VTA DA neurons is dominant. In contrast, if more CB1Rs are expressed in VTA or VTA-projecting glutamatergic neurons, cannabis will be aversive as CB1R mediated reduction in glutamatergic inputs onto VTA DA neurons is dominant. Congruently, if CB1R levels are equivalent on both types of neurons, cannabis should have no net effect on the brain reward function (Figure 2). This hypothesis appears to well explain why Δ9-THC or cannabinoids are rewarding in some human subjects and non-human primates (squirrel monkeys) in which more CB1Rs might be expressed in VTA-projecting GABA neurons but are ineffective or aversive in other human subjects and species (such as rats, mice) in which more CB1Rs might be expressed in VTA or VTA-projecting glutamate neurons (Figure 2).4.3. Dopaminergic CB2 Hypothesis of Cannabinoid Aversion
In addition to the above glutamate-CB1R hypothesis, recent research indicates that CB2Rs are also expressed in VTA DA neurons and contribute to the aversive effects of cannabinoids [3][82][3,82]. Activation of CB2Rs by JWH133 inhibits VTA DA neuron activity and decreases NAc DA release in wildtype mice, but not in CB2-KO mice [83][84][85][86][83,84,85,86]. Activation of CB2Rs or overexpression of brain CB2Rs also inhibits cocaine self-administration, cocaine-induced CPP and hyperactivity in mice [85][87][88][89][85,87,88,89]. In rats, Δ9-THC and WIN55, 212-2 produce biphasic effects on electrical brain-stimulation reward (BSR)—reward-enhancing at lower doses and reward-attenuating (or aversive) at higher doses [63]. CB1R antagonism (by AM251) reduced the low dose-enhanced BSR, while CB2R antagonism (AM630) decreased the high dose-attenuated BSR. Congruently, selective CB1R and CB2R agonists produced significant BSR enhancement and inhibition, respectively [63]. Together, these findings suggest that DA-CB2R mechanisms, at least in part, underlie cannabinoid-induced aversion (Figure 2) [3][23][3,23]. Thus, the subjective effects of cannabinoids would depend on the balance of multiple cell type-specific receptor mechanisms. This cell type-specific cannabinoid receptor mechanism appears to well explain why cannabis or cannabinoids could be rewarding, aversive, or ineffective since the cellular distributions of CB1Rs and CB2Rs may be different in different subjects or species.5. CB1R Antagonists Are Promising for the Treatments of SUDs
Rationale: In a series of clinical trials known as the Studies with Rimonabant and Tobacco Use (STRATUS), it was found that rimonabant significantly increased abstinence rates and reduced smoking cessation-related weight gain [9][90][91][92][93][9,112,113,114,115], suggesting that the endocannabinoid system may also be involved in nicotine use disorder and CB1R antagonists or inverse agonists may be useful for the treatment of SUDs including nicotine use disorder. Supporting evidence: The findings with either CB1R agonists or antagonists support this hypothesis of eCB involvement in SUDs. For example, Δ9-THC increases heroin self-administration in rats [94][116]. WIN55,212-2 increases motivation to nicotine self-administration and facilitates cue-induced reinstatement of nicotine seeking in rats [95][117]. WIN55,212-2 or CP55,940 facilitate alcohol self-administration, CPP, and binge-like behavior in rodents [96][97][118,119]. Accordingly, it was proposed that CB1R antagonists should also be effective in the treatment of SUDs [8][98][99][8,120,121]. Indeed, compelling preclinical evidence supports this hypothesis. For example, rimonabant reduced intravenous heroin self-administration under fixed-ratio and progressive-ratio reinforcement [100][101][122,123], nicotine cue-induced reinstatement of nicotine-seeking behavior in rats [102][103][124,125], and nicotine-enhanced DA release in the NAc [102][124]. Furthermore, rimonabant also prevents the development of morphine-induced CPP [104][126], and dose-dependently attenuates heroin- or heroin-associated cue-induced reinstatement of drug-seeking behavior [101][123]. In addition, rimonabant blocks acquisition of cocaine-induced CPP [105][127] and attenuates reinstatement of drug seeking caused by cocaine or cocaine-associated cues [106][128]). Congruently, rimonabant also blocks reinstatement of methamphetamine-seeking behavior [107][129] and reduces alcohol intake in rodents [108][130]. These exciting findings with rimonabant encouraged many pharmaceutical industries to develop other brain-penetrant CB1R antagonists such as the longer acting second generation surinabant (SR147778, Sanofi-Aventis), taranabant (MK-0364, Merck), otenabant (CP-945,598, Pfizer), and ibipinabant (SLV319, Solvay Pharmaceutical), for the treatment of obesity, smoking and drugs of abuse [109][102]. Receptor mechanisms: Unfortunately, all the above-mentioned ligands are not only CB1R antagonists but also inverse agonists. Thus, dissecting the role of CB1R antagonism versus inverse agonism in their therapeutic versus side-effects is critical for developing safer CB1R ligands for the treatment of SUDs. CB1R antagonism may underlie the therapeutic effects of rimonabant: Given that rimonabant is a well-characterized CB1R antagonist, it is reasonable to hypothesize that the CB1R antagonism may underlie its therapeutic anti-obesity and anti-addictive effects. This is supported by several lines of evidence. First, cannabinoids and drugs of abuse may act in a common neural substrate—the mesocorticolimbic DA system via distinct cellular and receptor mechanisms (Figure 2). Assuming that CB1Rs are tonically activated by eCBs (2-AG, AEA), CB1R antagonism on GABA neurons or GABAergic terminals would produce a reduction in eCB-enhanced DA transmission (Figure 2), which may functionally counteract the DA-enhancing and the pro-addictive effects produced by drugs of abuse. Second, growing evidence indicates that drugs of abuse (such as cocaine, heroin, or nicotine) may increase eCB release in the VTA and/or NAc [110][111][112][113][114][115][116][117][131,132,133,134,135,136,137,138] (Figure 3). After releasing from post-synaptic neurons, such as VTA DA neurons (Wang et al., 2015), eCBs retrogradely diffuse back to activate presynaptic CB1Rs, producing a reduction in neurotransmitter (GABA, glutamate) release, which subsequently produces reward-enhancing and addictive effects (Figure 3). Accordingly, CB1R antagonism at presynaptic terminals would block the eCB-mediated effects, producing anti-addictive effects (Figure 3). Third, the neutral CB1R antagonists without CB1R inverse agonist profile (such as AM4113 and PIMSR) produce the similar anti-addictive effects as rimonabant in drug self-administration and reinstatement, but without rimonabant-like depressive effects [7][13][118][7,13,139]. Lastly, in theory, rimonabant’s anhedonic or aversive effects may also functionally counteract the rewarding effects of drug abuse; however, direct supporting evidence is missing due to the absence of selective CB1R inverse agonists.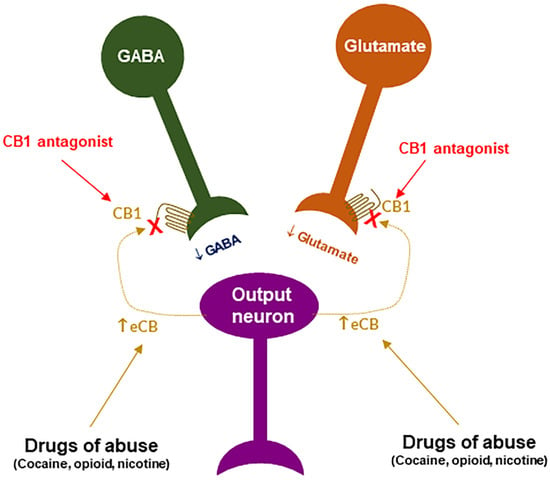
Figure 3. A schematic diagram of an endocannabinoid (eCB) hypothesis showing how a CB1R antagonist blocks actions produced by drugs of abuse. Growing evidence indicates that drugs of abuse (such as cocaine, opioid or nicotine) may stimulate endocannabinoid (such as 2-AG, AEA) release from postsynaptic neurons (such as VTA DA neurons), which subsequently activates CB1R on presynaptic CB1Rs and produces a reduction in GABA or glutamate release. A CB1R antagonist, including neutral antagonist, binds to CB1R and blocks eCB binding to the same receptor, therefore producing antagonism of drug reward and relapse. ↑: indicates an increase in eCB release; “×” indicates blockade of CB1 receptor.
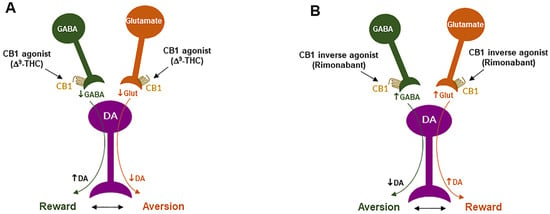
Figure 4. Schematic diagrams of a working hypothesis showing how a CB1R agonist and an inverse agonist produce opposite subjective effects. (A): A CB1R agonist such as Δ9-THC may produce rewarding effects by decreasing VTA GABA release and increasing NAc DA release. (B): In contrast, an inverse CB1R agonist may produce an opposite aversive effect by increasing GABA release and decreasing DA release in the NAc. ↑, ↓: indicate an increase or a decrease in neurotransmitter release.
Table 1. The literature reports regarding the rewarding vs. aversive effects of SR141716 itself in rodents.
| Compound | Doses | Species | Results | References | |
|---|---|---|---|---|---|
| Rimonabant | 20 mg/kg, i.p. | Rat | ↓ Electrical brain-stimulation reward | [121] | [142] |
| Rimonabant | 0.3, 1, 3, 10 mg/kg, i.p. | Rat | ↓ Electrical brain-stimulation reward | [118] | [139] |
| Rimonabant | 0.02, 0.3, 1.0 mg/kg, i.p. | Rat | No effect on electrical brain-stimulation reward | [123] | [144] |
| Rimonabant | 0.02 mg/kg, i.p. | Rat | No effect on electrical brain-stimulation reward | [124] | [145] |
| Rimonabant | 3, 10 mg/kg, i.p., | Mouse | No effect on electrical brain-stimulation reward | [122] | [143] |
| Rimonabant | 0.3, 1, 3 mg/kg, i.p. | Rat | Not produce CPP or CPA | [105] | [127] |
| Rimonabant | 0.1, 0.5, 3.0 mg/kg, i.p. | Rat | Not produces CPP or CPA | [127] | [148] |
| Rimonabant | 0.5, 1, 2 mg/kg, i.p. | Rat | Not produce CPP or CPA | [128] | [149] |
| Rimonabant | 3 mg/kg, i.p. | Rat | Not produce CPP or CPA | [89] | |
| Rimonabant | 0.25, 0.5, 1 mg/kg, i.p. | Rat | Not produce CPP or CPA | [55] | |
| Rimonabant | 0.1, 0.5, 3 mg/kg | Rat | Not produce CPP or CPA | [129] | [150] |
| Rimonabant | 0.3, 3 mg/kg, i.p. | Rat | Not produce CPP or CPA | [130] | [151] |
| Rimonabant | 0.25, 0.5, 2, 3 mg/kg | Rat | Produces CPP | [53] | |
| Rimonabant | 3 mg/kg | Mouse | ↓ Accumbens DA | [125] | [146] |
| Rimonabant | 2, 10 mg/kg, i.p. | Rat | No effect on accumbens DA | [76] | |
| Rimonabant | 1, 10, 30, 100 mM, intra-NAc | Rat | ↑ Accumbens DA | [76] | |
| Rimonabant | 5, 10, 20 mg/kg, i.p. | Rat | ↑ Accumbens DA | [126] | [147] |