 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Joana M. Gaspar | -- | 2671 | 2023-01-19 13:15:17 | | | |
| 2 | Peter Tang | Meta information modification | 2671 | 2023-01-20 02:38:39 | | |
Video Upload Options
Obesity is defined as abnormal and excessive fat accumulation, and it is a risk factor for developing metabolic and neurodegenerative diseases and cognitive deficits. Obesity is caused by an imbalance in energy homeostasis resulting from increased caloric intake associated with a sedentary lifestyle. However, the entire physiopathology linking obesity with neurodegeneration and cognitive decline has not yet been elucidated. During the progression of obesity, adipose tissue undergoes immune, metabolic, and functional changes that induce chronic low-grade inflammation. It has been proposed that inflammatory processes may participate in both the peripheral disorders and brain disorders associated with obesity, including the development of cognitive deficits. In addition, mitochondrial dysfunction is related to inflammation and oxidative stress, causing cellular oxidative damage.
1. Introduction
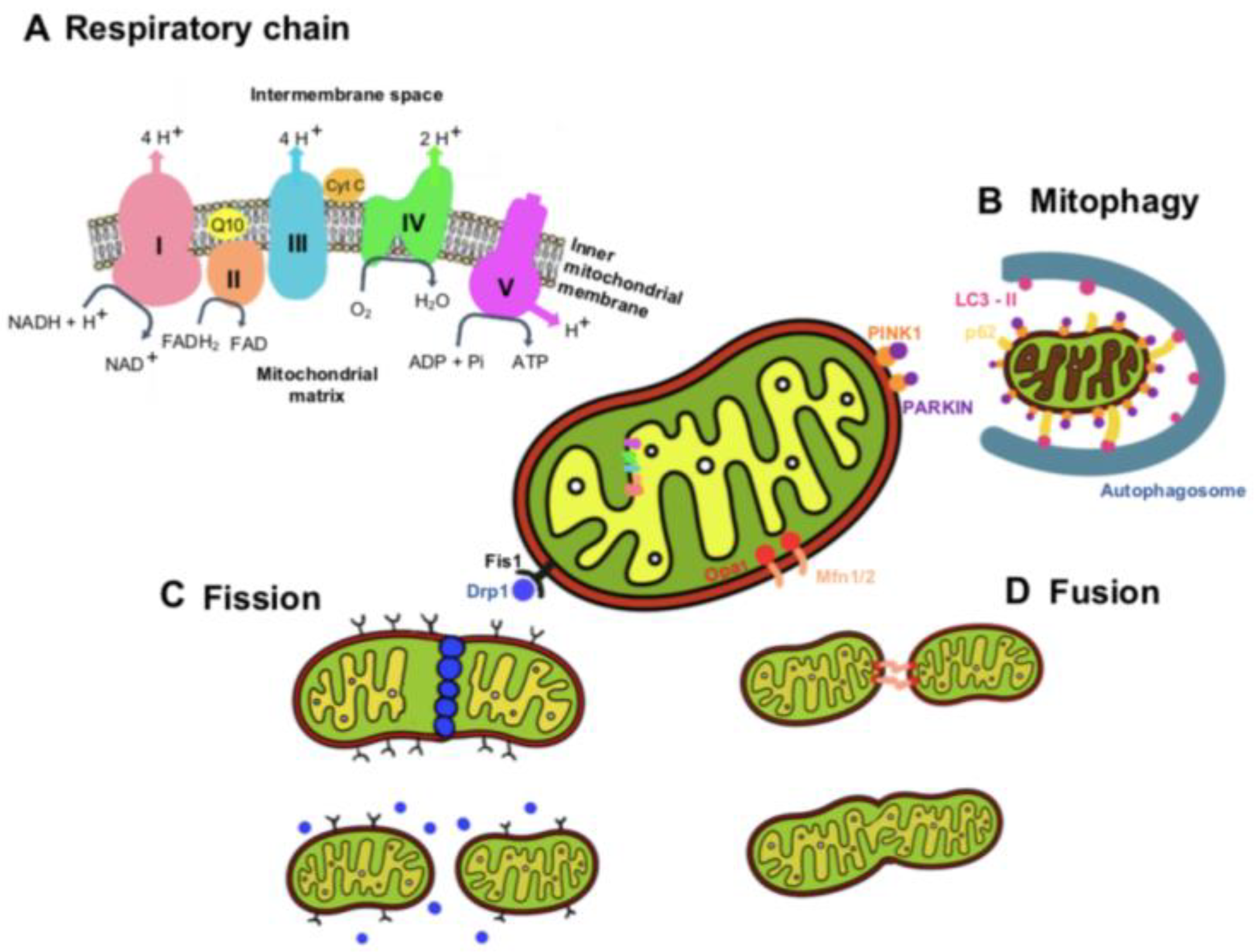
2. Mitochondria Functions and Dynamics
3. Obesity Induces Cognitive Decline
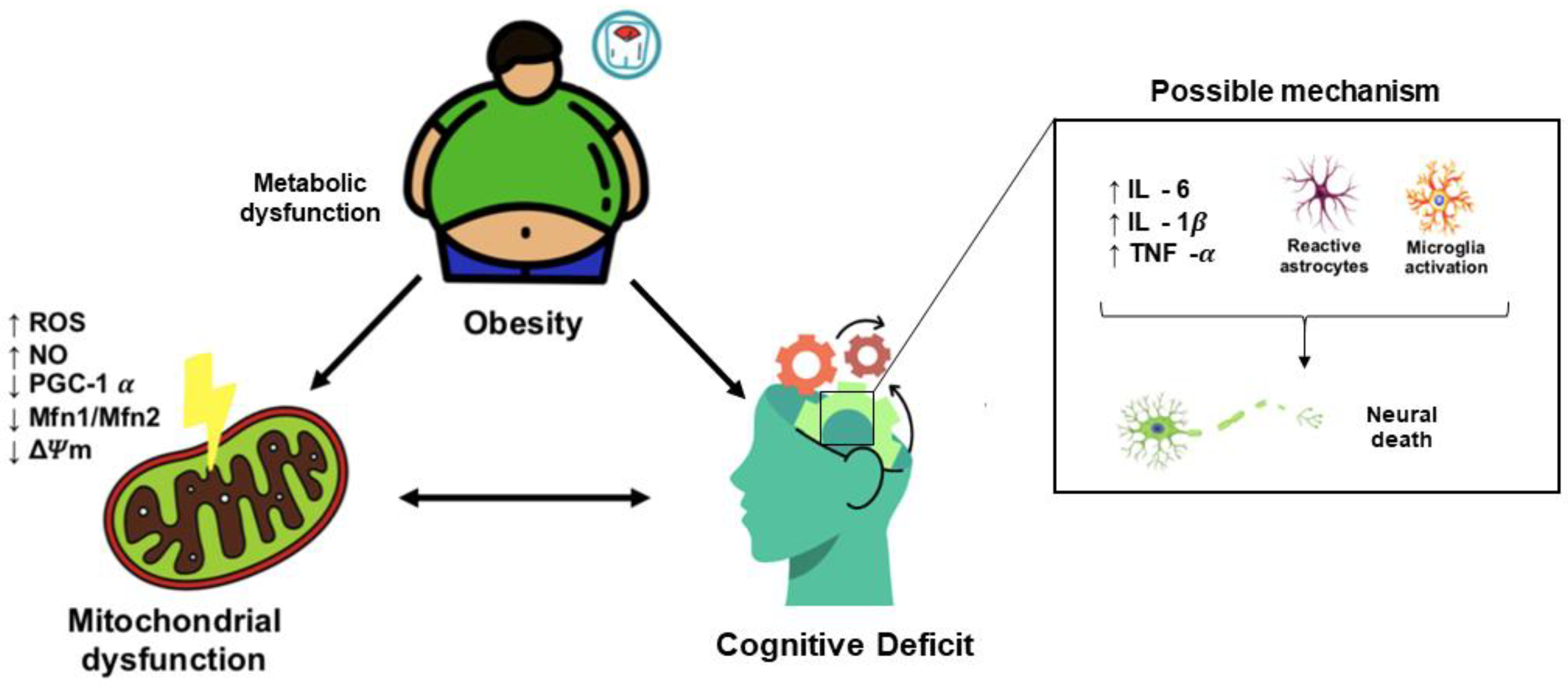
3.1. Obesity-Induced Cognitive Decline: Role of Neuroinflammation
3.2. Obesity-Induced Cognitive Decline: Role of Mitochondrial Dysfunction
References
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298.
- Collaborators, G.B.D.O.; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27.
- Morys, F.; Dadar, M.; Dagher, A. Association Between Midlife Obesity and Its Metabolic Consequences, Cerebrovascular Disease, and Cognitive Decline. J. Clin. Endocrinol. Metab. 2021, 106, e4260–e4274.
- De Lorenzo, A.; Gratteri, S.; Gualtieri, P. Why primary obesity is a disease? J. Transl. Med. 2019, 17, 169.
- Pratchayasakul, W.; Kerdphoo, S.; Petsophonsakul, P.; Pongchaidecha, A.; Chattipakorn, N.; Chattipakorn, S. Effects of high-fat diet on insulin receptor function in rat hippocampus and the level of neuronal corticosterone. Life Sci. 2011, 88, 619–627.
- Pipatpiboon, N.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. PPARγ agonist improves neuronal insulin receptor function in hippocampus and brain mitochondria function in rats with insulin resistance induced by long term high-fat diets. Endocrinology 2012, 153, 329–338.
- Abbasnejad, Z.; Nasseri, B.; Zardooz, H.; Ghasemi, R. Time-course study of high fat diet induced alterations in spatial memory, hippocampal JNK, P38, ERK and Akt activity. Metab. Brain Dis. 2019, 34, 659–673.
- Gómez-Apo, E.; Mondragón-Maya, A.; Ferrari-Díaz, M.; Silva-Pereyra, J. Structural Brain Changes Associated with Overweight and Obesity. J. Obes. 2021, 2021, 6613385.
- Gaspar, J.M.; Baptista, F.I.; Macedo, M.P.; Ambrósio, A.F. Inside the Diabetic Brain: Role of Different Players Involved in Cognitive Decline. ACS Chem. Neurosci. 2016, 7, 131–142.
- Gispen, W.H.; Biessels, G.J. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000, 23, 542–549.
- Fitzpatrick, A.L.; Kuller, L.H.; Lopez, O.L.; Diehr, P.; O’Meara, E.S.; Longstreth, W.T., Jr.; Luchsinger, J.A. Midlife and late-life obesity and the risk of dementia: Cardiovascular health study. Arch. Neurol. 2009, 66, 336–342.
- Hughes, T.F.; Borenstein, A.R.; Schofield, E.; Wu, Y.; Larson, E.B. Association between late-life body mass index and dementia: The Kame Project. Neurology 2009, 72, 1741–1746.
- Whitmer, R.A.; Gunderson, E.P.; Quesenberry, C.P., Jr.; Zhou, J.; Yaffe, K. Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Curr. Alzheimer Res. 2007, 4, 103–109.
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet. Neurol. 2006, 5, 64–74.
- Lu, F.P.; Lin, K.P.; Kuo, H.K. Diabetes and the risk of multi-system aging phenotypes: A systematic review and meta-analysis. PLoS ONE 2009, 4, e4144.
- Marseglia, A.; Darin-Mattsson, A.; Skoog, J.; Rydén, L.; Hadarsson-Bodin, T.; Kern, S.; Rydberg Sterner, T.; Shang, Y.; Zettergren, A.; Westman, E.; et al. Metabolic Syndrome Is Associated With Poor Cognition: A Population-Based Study of 70-Year-Old Adults Without Dementia. J. Gerontol. Ser. A 2021, 76, 2275–2283.
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224.
- Bras, M.; Queenan, B.; Susin, S.A. Programmed cell death via mitochondria: Different modes of dying. Biochemistry 2005, 70, 231–239.
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism. Nature 1961, 191, 144–148.
- Hatefi, Y. The Mitochondrial Electron Transport and Oxidative Phosphorylation System. Annu. Rev. Biochem. 1985, 54, 1015–1069.
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15.
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674.
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259.
- Galizzi, G.; Di Carlo, M. Insulin and Its Key Role for Mitochondrial Function/Dysfunction and Quality Control: A Shared Link between Dysmetabolism and Neurodegeneration. Biology 2022, 11, 943.
- Cardoso, S.; López, I.P.; Piñeiro-Hermida, S.; Pichel, J.G.; Moreira, P.I. IGF1R Deficiency Modulates Brain Signaling Pathways and Disturbs Mitochondria and Redox Homeostasis. Biomedicines 2021, 9, 158.
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free. Radic. Biol. Med. 2014, 74, 129–144.
- Cheng, Y.; Buchan, M.; Vitanova, K.; Aitken, L.; Gunn-Moore, F.J.; Ramsay, R.R.; Doherty, G. Neuroprotective actions of leptin facilitated through balancing mitochondrial morphology and improving mitochondrial function. J. Neurochem. 2020, 155, 191–206.
- Jong, C.J.; Yeung, J.; Tseung, E.; Karmazyn, M. Leptin-induced cardiomyocyte hypertrophy is associated with enhanced mitochondrial fission. Mol. Cell. Biochem. 2019, 454, 33–44.
- Yang, F.; Li, B.; Yang, Y.; Huang, M.; Liu, X.; Zhang, Y.; Liu, H.; Zhang, L.; Pan, Y.; Tian, S.; et al. Leptin enhances glycolysis via OPA1-mediated mitochondrial fusion to promote mesenchymal stem cell survival. Int. J. Mol. Med. 2019, 44, 301–312.
- Wauman, J.; Tavernier, J. The intracellular domain of the leptin receptor prevents mitochondrial depolarization and mitophagy. Biochim. Biophys. Acta. Mol. Cell Res. 2018, 1865, 1312–1325.
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158.
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667.
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200.
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862.
- Qi, Y.; Yan, L.; Yu, C.; Guo, X. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 2016, 215, 621–629.
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532.
- Ward, M.A.; Carlsson, C.M.; Trivedi, M.A.; Sager, M.A.; Johnson, S.C. The effect of body mass index on global brain volume in middle-aged adults: A cross sectional study. BMC Neurol. 2005, 5, 23.
- Haltia, L.T.; Viljanen, A.; Parkkola, R.; Kemppainen, N.; Rinne, J.O.; Nuutila, P.; Kaasinen, V. Brain white matter expansion in human obesity and the recovering effect of dieting. J. Clin. Endocrinol. Metab. 2007, 92, 3278–3284.
- Gazdzinski, S.; Kornak, J.; Weiner, M.W.; Meyerhoff, D.J. Body mass index and magnetic resonance markers of brain integrity in adults. Ann. Neurol. 2008, 63, 652–657.
- Chen, R.; Cai, G.; Xu, S.; Sun, Q.; Luo, J.; Wang, Y.; Li, M.; Lin, H.; Liu, J. Body mass index related to executive function and hippocampal subregion volume in subjective cognitive decline. Front. Aging Neurosci. 2022, 14, 905035.
- Kharabian Masouleh, S.; Arélin, K.; Horstmann, A.; Lampe, L.; Kipping, J.A.; Luck, T.; Riedel-Heller, S.G.; Schroeter, M.L.; Stumvoll, M.; Villringer, A.; et al. Higher body mass index in older adults is associated with lower gray matter volume: Implications for memory performance. Neurobiol. Aging 2016, 40, 1–10.
- Gustafson, D.; Rothenberg, E.; Blennow, K.; Steen, B.; Skoog, I. An 18-year follow-up of overweight and risk of Alzheimer disease. Arch. Intern. Med. 2003, 163, 1524–1528.
- Dahl, A.; Hassing, L.B.; Fransson, E.; Berg, S.; Gatz, M.; Reynolds, C.A.; Pedersen, N.L. Being overweight in midlife is associated with lower cognitive ability and steeper cognitive decline in late life. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65, 57–62.
- Gunstad, J.; Lhotsky, A.; Wendell, C.R.; Ferrucci, L.; Zonderman, A.B. Longitudinal examination of obesity and cognitive function: Results from the Baltimore longitudinal study of aging. Neuroepidemiology 2010, 34, 222–229.
- Gunstad, J.; Paul, R.H.; Cohen, R.A.; Tate, D.F.; Spitznagel, M.B.; Gordon, E. Elevated body mass index is associated with executive dysfunction in otherwise healthy adults. Compr. Psychiatry 2007, 48, 57–61.
- Quaye, E.; Galecki, A.T.; Tilton, N.; Whitney, R.; Briceño, E.M.; Elkind, M.S.V. Association of Obesity with Cognitive Decline in Black and White Americans. Neurology 2022.
- Baym, C.L.; Khan, N.A.; Monti, J.M.; Raine, L.B.; Drollette, E.S.; Moore, R.D.; Scudder, M.R.; Kramer, A.F.; Hillman, C.H.; Cohen, N.J. Dietary lipids are differentially associated with hippocampal-dependent relational memory in prepubescent children. Am. J. Clin. Nutr. 2014, 99, 1026–1032.
- Francis, H.M.; Stevenson, R.J. Higher reported saturated fat and refined sugar intake is associated with reduced hippocampal-dependent memory and sensitivity to interoceptive signals. Behav. Neurosci. 2011, 125, 943–955.
- Attuquayefio, T.; Stevenson, R.J.; Oaten, M.J.; Francis, H.M. A four-day Western-style dietary intervention causes reductions in hippocampal-dependent learning and memory and interoceptive sensitivity. PLoS ONE 2017, 12, e0172645.
- Wolf, P.A.; Beiser, A.; Elias, M.F.; Au, R.; Vasan, R.S.; Seshadri, S. Relation of obesity to cognitive function: Importance of central obesity and synergistic influence of concomitant hypertension. The Framingham Heart Study. Curr. Alzheimer Res. 2007, 4, 111–116.
- Snyder, L.L.; Foland-Ross, L.C.; Cato, A.; Reiss, A.L.; Shah, C.; Hossain, J.; Elmufti, H.; Nelly, M. Impact of dysglycemia and obesity on the brain in adolescents with and without type 2 diabetes: A pilot study. Pediatr. Diabetes 2022, 23, 1674–1686.
- Cserjési, R.; Molnár, D.; Luminet, O.; Lénárd, L. Is there any relationship between obesity and mental flexibility in children? Appetite 2007, 49, 675–678.
- Liang, J.; Matheson, B.E.; Kaye, W.H.; Boutelle, K.N. Neurocognitive correlates of obesity and obesity-related behaviors in children and adolescents. Int. J. Obes. 2014, 38, 494–506.
- Beeri, M.S.; Tirosh, A.; Lin, H.M.; Golan, S.; Boccara, E.; Sano, M.; Zhu, C.W. Stability in BMI over time is associated with a better cognitive trajectory in older adults. Alzheimer’s Dement. 2022, 18, 2131–2139.
- Li, C.M.; Song, J.R.; Zhao, J.; Wang, C.F.; Zhang, C.S.; Wang, H.D.; Zhang, Q.; Liu, D.F.; Ma, Z.Y.; Yuan, J.H.; et al. The effects of bariatric surgery on cognition in patients with obesity: A systematic review and meta-analysis. Surg. Obes. Relat. Dis. 2022, 18, 1323–1338.
- Dardano, A.; Aghakhanyan, G.; Moretto, C.; Ciccarone, A.; Bellini, R.; Ceccarini, G.; Sancho Bornez, V.; Santini, F.; Volterrani, D.; Del Prato, S.; et al. Brain effect of bariatric surgery in people with obesity. Int. J. Obes. 2022, 46, 1671–1677.
- de Paula, G.C.; Brunetta, H.S.; Engel, D.F.; Gaspar, J.M.; Velloso, L.A.; Engblom, D.; de Oliveira, J.; de Bem, A.F. Hippocampal Function Is Impaired by a Short-Term High-Fat Diet in Mice: Increased Blood-Brain Barrier Permeability and Neuroinflammation as Triggering Events. Front. Neurosci. 2021, 15, 734158.
- Hao, S.; Dey, A.; Yu, X.; Stranahan, A.M. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav. Immun. 2016, 51, 230–239.
- Hsu, T.M.; Konanur, V.R.; Taing, L.; Usui, R.; Kayser, B.D.; Goran, M.I.; Kanoski, S.E. Effects of sucrose and high fructose corn syrup consumption on spatial memory function and hippocampal neuroinflammation in adolescent rats. Hippocampus 2015, 25, 227–239.
- Stranahan, A.M.; Arumugam, T.V.; Cutler, R.G.; Lee, K.; Egan, J.M.; Mattson, M.P. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat. Neurosci. 2008, 11, 309–317.
- Boitard, C.; Cavaroc, A.; Sauvant, J.; Aubert, A.; Castanon, N.; Layé, S.; Ferreira, G. Impairment of hippocampal-dependent memory induced by juvenile high-fat diet intake is associated with enhanced hippocampal inflammation in rats. Brain Behav. Immun. 2014, 40, 9–17.
- Lizarbe, B.; Soares, A.F.; Larsson, S.; Duarte, J.M.N. Neurochemical Modifications in the Hippocampus, Cortex and Hypothalamus of Mice Exposed to Long-Term High-Fat Diet. Front. Neurosci. 2018, 12, 985.
- Howe, A.M.; Burke, S.; O’Reilly, M.E.; McGillicuddy, F.C.; Costello, D.A. Palmitic Acid and Oleic Acid Differently Modulate TLR2-Mediated Inflammatory Responses in Microglia and Macrophages. Mol. Neurobiol. 2022, 59, 2348–2362.
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014, 9, 2124–2138.
- Carraro, R.S.; Souza, G.F.; Solon, C.; Razolli, D.S.; Chausse, B.; Barbizan, R.; Victorio, S.C.; Velloso, L.A. Hypothalamic mitochondrial abnormalities occur downstream of inflammation in diet-induced obesity. Mol. Cell. Endocrinol. 2017, 460, 238–245.
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and TNF-alpha on appetite-regulating NPY neurons. Int. J. Obes. 2017, 41, 149–158.
- Gaspar, J.M.; Castilho, Á.; Baptista, F.I.; Liberal, J.; Ambrósio, A.F. Long-term exposure to high glucose induces changes in the content and distribution of some exocytotic proteins in cultured hippocampal neurons. Neuroscience 2010, 171, 981–992.
- Beilharz, J.E.; Maniam, J.; Morris, M.J. Short exposure to a diet rich in both fat and sugar or sugar alone impairs place, but not object recognition memory in rats. Brain Behav. Immun. 2014, 37, 134–141.
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830.
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91.
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415.
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282.
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162.
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199.
- Yu, G.; Cao, F.; Hou, T.; Cheng, Y.; Jia, B.; Yu, L.; Chen, W.; Xu, Y.; Chen, M.; Wang, Y. Astrocyte reactivation in medial prefrontal cortex contributes to obesity-promoted depressive-like behaviors. J. Neuroinflammation 2022, 19, 166.
- Salas-Venegas, V.; Flores-Torres, R.P.; Rodríguez-Cortés, Y.M.; Rodríguez-Retana, D.; Ramírez-Carreto, R.J.; Concepción-Carrillo, L.E.; Pérez-Flores, L.J.; Alarcón-Aguilar, A.; López-Díazguerrero, N.E.; Gómez-González, B.; et al. The Obese Brain: Mechanisms of Systemic and Local Inflammation, and Interventions to Reverse the Cognitive Deficit. Front. Integr. Neurosci. 2022, 16, 798995.
- Guo, D.-H.; Yamamoto, M.; Hernandez, C.M.; Khodadadi, H.; Baban, B.; Stranahan, A.M. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J. Clin. Investig. 2020, 130, 1961–1976.
- Buckman, L.B.; Hasty, A.H.; Flaherty, D.K.; Buckman, C.T.; Thompson, M.M.; Matlock, B.K.; Weller, K.; Ellacott, K.L. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav. Immun. 2014, 35, 33–42.
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73.
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; François, P.; de Vos, W.M.; et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 2011, 60, 2775–2786.
- Bruce-Keller, A.J.; Salbaum, J.M.; Luo, M.; Blanchard, E.; Taylor, C.M.; Welsh, D.A.; Berthoud, H.R. Obese-type gut microbiota induce neurobehavioral changes in the absence of obesity. Biol. Psychiatry 2015, 77, 607–615.
- Wardzinski, E.K.; Kistenmacher, A.; Melchert, U.H.; Jauch-Chara, K.; Oltmanns, K.M. Impaired brain energy gain upon a glucose load in obesity. Metabolism 2018, 85, 90–96.
- Jayashankar, V.; Selwan, E.; Hancock, S.E.; Verlande, A.; Goodson, M.O.; Eckenstein, K.H.; Milinkeviciute, G.; Hoover, B.M.; Chen, B.; Fleischman, A.G.; et al. Drug-like sphingolipid SH-BC-893 opposes ceramide-induced mitochondrial fission and corrects diet-induced obesity. EMBO Mol. Med. 2021, 13, e13086.


