Obesity is a chronic disease defined as abnormal and excessive fat accumulation, and it represents an important risk factor for many diseases and premature death. Body mass index (BMI) is a weight-for-height index commonly used to classify overweight and obesity in adults. The healthy weight range is a BMI range between 18.5 and <24.9 kg/m
2. If an individual’s BMI is between 25.0 and 29.9 kg/m
2, they fall within the overweight range; if their BMI is 30.0 kg/m
2 or higher, they are considered obese. In 2015, more than 1.9 billion adults were overweight; over 600 million were obese [
1,
2]. Even more alarming is that child obesity affects 107.7 million children [
1,
2]. Changes in lifestyle in the last century (increased consumption of hypercaloric diets and sedentary behavior) are the fundamental causes of obesity epidemics.
2. Mitochondria Functions and Dynamics
Mitochondria are double-membrane organelles responsible for energy production and homeostasis, the regulation of intracellular calcium levels, and the regulation of apoptosis (mainly via the intrinsic pathway) [
17,
18]. In addition, mitochondria are responsible for generating more than 90% of the energy for the cell through oxidative phosphorylation [
19,
20].
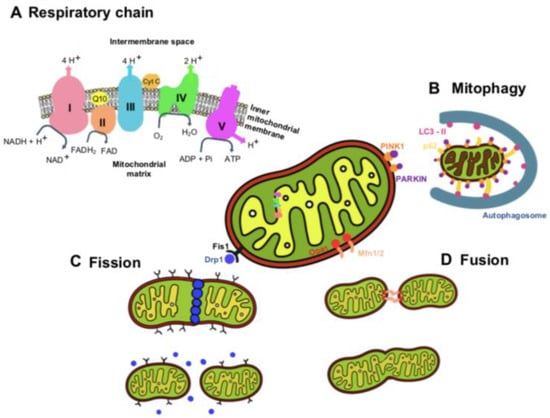
To generate ATP through oxidative phosphorylation, mitochondria use an electron transport chain inserted within the mitochondrion’s inner membrane (
Figure 1) [
21]. NADH and FADH
2 are generated by the Krebs cycle and donate electrons to complex I (NADH: ubiquinone oxidoreductase) and complex II (succinate dehydrogenase), respectively. The electrons from NADH are passed from complex I to ubiquinone (CoQ) in order to enter the Q cycle, where CoQ is reduced to ubiquinol (QH
2). This electron transfer induces the pumping of protons by complex I from the matrix into the intermembrane space. The electrons donated from FADH
2 are transferred from complex II to CoQ similarly to complex I, although this process is not accompanied by proton translocation [
21]. Once in the Q cycle, the electrons are transferred to complex III (coenzyme Q: cytochrome c reductase) and then to cytochrome c, releasing two protons into the intermembrane space. Then, when cytochrome c is reduced, it transports single electrons from complex III to complex IV (cytochrome c oxidase), where molecular oxygen is reduced to water. At complex IV, a total of eight protons are pumped from the matrix, of which four are used to form two water molecules, and the other four are transferred into the intermembrane space [
21,
22].
In response to electron transport, a total of ten protons are pumped from the matrix into the intermembrane space, where they accumulate to generate an electrochemical and concentration proton gradient that generates a proton motive force, essential for the activity of complex V (ATP synthase) to generate ATP [
21]. A consequence of electron transfer is the generation of reactive oxygen species (ROS), which contributes to homeostatic signaling. However, when ROS are produced in excess, they cause oxidative stress and can lead to mitochondrial dysfunction and diseases [
22]. Therefore, an efficient measurement of the electron transport chain function and ATP production, using high-resolution respirometry, such as a Seahorse XF24 Extracellular Flux Analyzer and oxygraphy, can provide insight into cellular physiology and dysfunction.
Mitochondria are highly dynamic organelles that undergo a continuous cycle of fission and fusion, processes called mitochondrial dynamics (
Figure 1). Another dynamic process of mitochondria is the selective removal of dysfunctional mitochondria, a quality-control mechanism that ensures a healthy mitochondrial population. The dynamic properties of mitochondria are critical for their optimal function in energy generation [
23]. Mitophagy is a mechanism of mitochondrial quality control used to eliminate damaged mitochondria and prevent excessive ROS production, thus maintaining homeostasis in mitochondria. Mitochondrial dynamics involve the plasma membrane and organelles, such as ER and lysosomes. The contact point of ER–mitochondria is referred to as mitochondria-associated ER membranes. Some studies have suggested that the integrity of mitochondria-associated ER membranes is required for insulin signaling (for a detailed description, see the revision [
24]). Studies have been carried out to investigate the effect/defect of insulin signaling on different features of mitochondrial dysfunction, focusing on dynamics, biogenesis, and mitophagy and their role in pathologies in which metabolic dysmetabolism is comorbid with neurodegeneration [
25,
26]. Some studies have also suggested that the protective actions of leptin may be facilitated through the regulation of mitochondrial dynamics, namely, mitochondrial fission and fusion [
27,
28,
29,
30].
Dysfunctional mitochondria are recognized by the autophagy machinery, resulting in their engulfment by autophagosomes and trafficking to the lysosome for degradation. The most common mitophagy pathways are mediated by PINK1 and PARKIN proteins. Mitochondrial fission is the process where mitochondria divide into two separate mitochondrial organelles. Fission is mediated by the interaction between the mitochondrial fission factor (Mff) and dynamin-related protein-1 (Drp1). Briefly, Drp1 is recruited from a cytosolic pool onto the mitochondrial surface, where it self-assembles into spiral structures to facilitate fission, acting similarly to endocytic invaginations of the cell membrane. Several mitochondrial-bound proteins then aid in the recruitment of Drp1 to the mitochondria, including Fis1, Mff, MiD49, and MiD51 [
23,
31,
32].
Fusion is the process of joining two adjacent mitochondria through a physical merging of the outer and then the inner mitochondrial membranes, resulting in the content mixing of the matrix components diffusing throughout the new mitochondrion. Fusion is mediated by the proteins mitofusin-1 (Mfn1) and mitofusin-2 (Mfn2) [
33,
34,
35], located on the mitochondrial outer membrane. Mitofusins are required for outer membrane fusion. The fusion of the inner membrane is mediated by the protein optic atrophy 1 (Opa1), which is associated with the inner membrane ([
36] for a detailed description of the mitochondria dynamics, please read the review manuscript [
23]).
Mitochondrial dynamics is important for growth redistribution and maintenance in a healthy mitochondria network and plays a role in disease-related processes. All the cells consume energy for their homeostasis and specific activity, and they require the support of functional mitochondria that provide ATP obtained via oxidative phosphorylation. A reduction in mitochondria respiration and bioenergetics is associated with insulin resistance [
24].
Therefore, the dysfunction of mitochondrial dynamics and function could lead to disorders in mitochondria, which are greatly associated with the progression of several diseases, including obesity and metabolic and neurological conditions.
3. Obesity Induces Cognitive Decline
Obesity, as well as HFD diet consumption, and metabolic disorders, such as diabetes mellitus, are widely recognized as inducing impairments in brain structure and function in the form of memory dysfunction, as well as neurodegenerative diseases [
37]. Furthermore, magnetic resonance imaging studies have demonstrated that regional brain atrophy and changes in gray and white matter are observed in patients with obesity, providing new insights into the relationship between obesity and cognitive decline from the imaging perspective [
37,
38,
39,
40]. Furthermore, a higher BMI is correlated with a lower gray matter volume in the prefrontal, temporal, insular, and occipital cortexes; thalamus; putamen; amygdala; and cerebellum, mediating the negative effects on memory performance [
41].
Patients with obesity have an earlier onset of Alzheimer’s, which is considered an aging disease [
42]. An 18-year follow-up longitudinal study demonstrated a higher degree of overweight in older women who developed AD. No associations were found in men [
42]. In the same study, the authors concluded that Alzheimer’s disease risk increased by 36% for every 1.0 increase in BMI. In other studies, it has been shown that patients with a higher BMI present significantly lower scores in cognitive tests and a longitudinal decline in cognitive abilities in both men and women [
37,
43,
44]. Changes in cognitive function can be potentiated since middle-aged adults with obesity may experience differentially greater brain atrophy [
37]. The relationship between a higher BMI and reduced cognitive performance does not change with age [
45] or race [
46]. A high intake of fat and sugar is associated with impairments in hippocampal-dependent learning and memory in children [
47] and adults [
48,
49], suggesting a negative impact on hippocampal function across the lifespan. In the community-based Framingham Offspring Cohort, it was observed that central obesity was significantly related to poorer performance in executive function and visuomotor skills, and no changes were observed for verbal memory [
50]. Adults with overweight and obesity also have poorer executive function than normal-weight adults, without changes in performance on attention tests. Children and adolescents with overweight/obesity also present poor cognitive function on verbal, full-scale, and performance IQ; visual–spatial; and executive function tests [
51,
52]. A systemic review found that executive dysfunction is associated with obesity-related behaviors in children and adolescents, such as increased food intake, disinhibited eating, and less physical activity. In children and adolescents, obesity is associated with poorer cognitive competence and may affect their academic achievements [
53].
Body weight and diet composition are modified risk factors for cognitive decline. Weight loss appears to be associated with low-order improvements in executive/attention functioning and memory in individuals with obesity. Moreover, a stable BMI predicts better cognitive trajectories [
54]. Patients with severe obesity may obtain immediate verbal and delayed memory function benefits from Roux-en-Y gastric bypass [
55,
56].
Different animal models of obesity and metabolic disorders have also exhibited cognitive dysfunctions and worse performance in learning and memory tasks compared to non-obese animals [
57,
58,
59,
60,
61,
62]. In addition, based on studies of animal models and in vitro models, high levels of glucose and saturated fatty acids are responsible for neuroinflammation, microglia activation, mitochondrial dysfunction, neuronal loss, and impairments in synaptic plasticity (
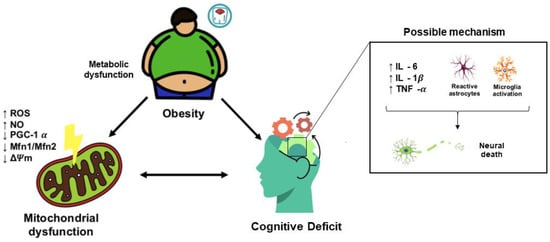
Figure 2) [
63,
64,
65,
66,
67,
68].