Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | ALESSANDRO Giammona | -- | 1643 | 2023-01-16 12:06:18 | | | |
| 2 | Conner Chen | Meta information modification | 1643 | 2023-01-17 07:07:56 | | | | |
| 3 | Conner Chen | + 2 word(s) | 1645 | 2023-01-18 09:06:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Giammona, A.; Crivaro, E.; Stecca, B. The Hedgehog Signaling Pathway. Encyclopedia. Available online: https://encyclopedia.pub/entry/40221 (accessed on 26 July 2026).
Giammona A, Crivaro E, Stecca B. The Hedgehog Signaling Pathway. Encyclopedia. Available at: https://encyclopedia.pub/entry/40221. Accessed July 26, 2026.
Giammona, Alessandro, Enrica Crivaro, Barbara Stecca. "The Hedgehog Signaling Pathway" Encyclopedia, https://encyclopedia.pub/entry/40221 (accessed July 26, 2026).
Giammona, A., Crivaro, E., & Stecca, B. (2023, January 16). The Hedgehog Signaling Pathway. In Encyclopedia. https://encyclopedia.pub/entry/40221
Giammona, Alessandro, et al. "The Hedgehog Signaling Pathway." Encyclopedia. Web. 16 January, 2023.
Copy Citation
Hedgehog–GLI (HH) signaling plays an essential role in embryogenesis and tissue homeostasis. Aberrant activation of the pathway through mutations or other mechanisms is involved in the development and progression of numerous types of cancer, including basal cell carcinoma, medulloblastoma, melanoma, breast, prostate, hepatocellular and pancreatic carcinomas. Activation of HH signaling sustains proliferation, suppresses cell death signals, enhances invasion and metastasis, deregulates cellular metabolism and promotes angiogenesis and tumor inflammation. Targeted inhibition of the HH pathway has therefore emerged as an attractive therapeutic strategy for the treatment of a wide range of cancers.
hedgehog signaling
tumor microenvironment
immunosuppression
1. Introduction
The Hedgehog–GLI (HH) signaling pathway plays key roles during embryonic development and is involved in cell proliferation, differentiation and tissue patterning. In adults, HH signaling is rapidly turned off and remains active in the stem cells of the central nervous system, skin and intestine, where it maintains tissue homeostasis and regeneration [1]. The HH signaling is aberrantly activated during the initiation and progression of a variety of cancer types, including those of the brain, skin, breast, prostate, hepatocellular and pancreatic carcinomas and hematological malignancies. The HH pathway is involved in enhancing proliferation, invasion and metastasis, in suppressing cell death signals and in deregulating the cellular metabolism [2]. Several reports have implicated HH signaling in suppressing the immune system and promoting an immunosuppressive tumor microenvironment (TME) [3]. Recent advances in cancer immunology and immunotherapy have emphasized the need for an accurate understanding of the immune-modulatory functions of oncogenic signaling pathways and their role in cancer immunity.
2. Hedgehog Signaling Pathway
The HH pathway is an evolutionary signaling pathway that plays a pivotal role in patterning and organogenesis during embryonic development and in adult tissue homeostasis and repair [1]. This complex transduction pathway is coordinated by several regulatory components and post-translational modifications. In mammals, HH signaling consists of three secreted HH ligands (Sonic Hedgehog, SHH; Desert Hedgehog, DHH; and Indian Hedgehog, IHH); the 12-pass transmembrane receptor Patched 1 (PTCH1); the 7-pass transmembrane G protein-coupled receptor (GPCR) Smoothened (SMO), as the main transducer of the HH pathway; and the three zinc finger GLI transcription factors (GLI1, GLI2, GLI3), as the final mediators of the transcriptional response of HH signaling [4]. Additional members include a number of regulatory kinases [5] and Suppressor of Fused (SUFU), the main negative regulator of the GLI [6] (Figure 1).
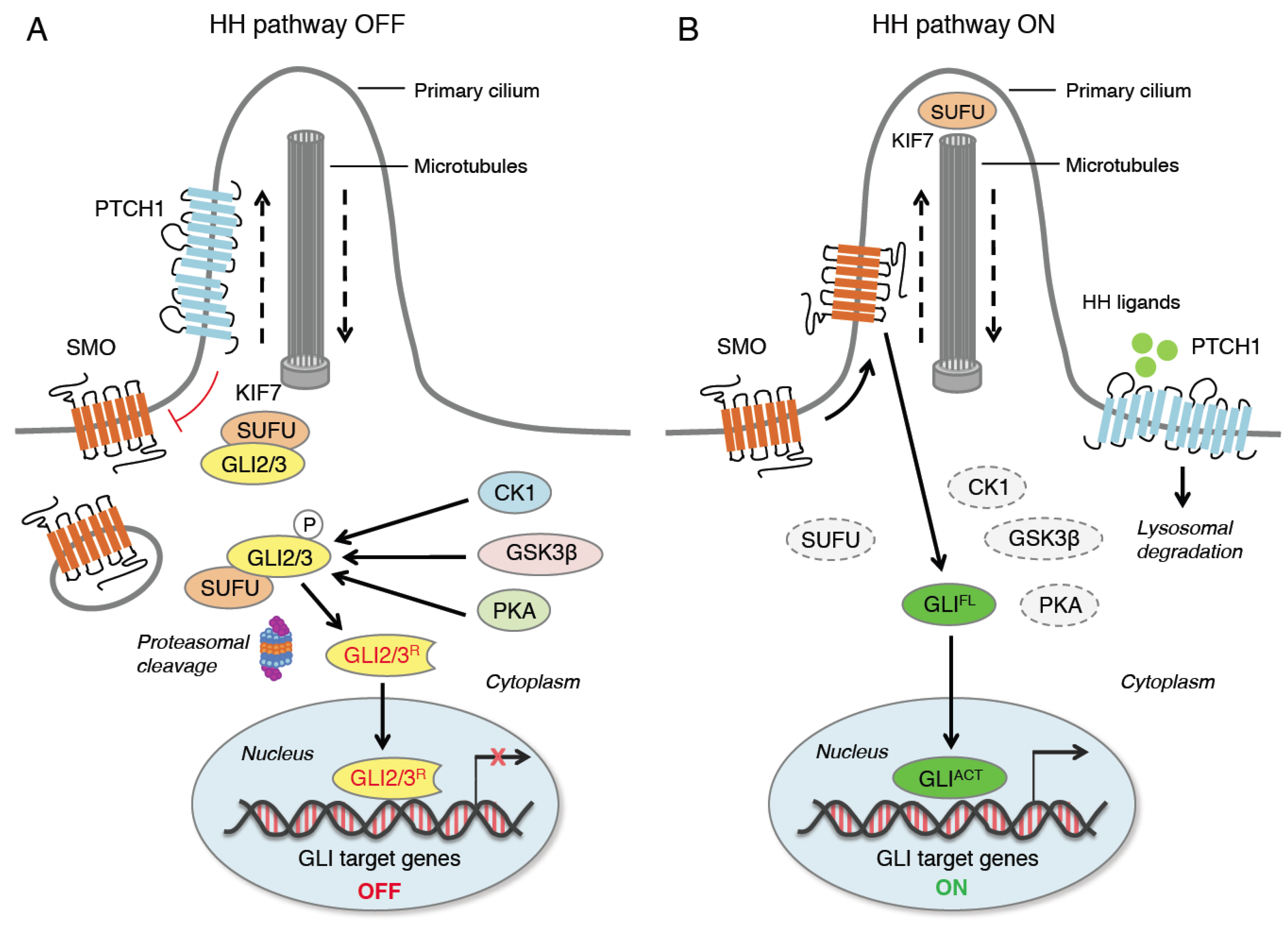
Figure 1. Canonical activation of HH signaling. When HH ligands are not present (A), PTCH1 represses SMO by preventing its entry into the primary cilium (PC). GLI2 and GLI3 are sequestered in the cytoplasm by SUFU and phosphorylated by PKA, CK1 and GSK3β. The GLI undergo ubiquitination through the E3 ubiquitin ligase β-TrCP. GLI1 is fully degraded, whereas GLI3 and, to a lesser extent, GLI2 undergo partial proteasome degradation, leading to the formation of repressor forms (GLI3/2R) that move into the nucleus inhibiting the transcription of GLI target genes. In the presence of HH ligands (B), PTCH1 is displaced from the PC and undergoes lysosomal degradation, and SMO translocates into the PC. Active SMO relieves the SUFU-mediated suppression of GLI2 and GLI3, triggering a signaling cascade that leads to the translocation of full length activated forms of GLI (GLIACT) into the nucleus, where they promote the transcription of GLI target genes. KIF7 is a kinesin protein that acts in anterograde transport (from base to tip) of the PC. CK1, casein kinase 1; GLI2/3R, GLI2/3 repressors; GLIACT, GLI activators; GLIFL, GLI full length; GSK3β, glycogen synthase kinase 3β; HH, Hedgehog; KIF7, kinesin family member 7; PKA, protein kinase A; PTCH1, Patched 1; SMO, Smoothened; SUFU, Suppressor of Fused; β-TrCP, β-transducin repeat-containing protein.
A simplified model of HH signaling proposes that in the absence of HH ligands PTCH1 localizes to the primary cilium (PC), an organelle specialized for HH pathway transduction [7], where it suppresses the ciliary accumulation of SMO. Therefore, GLI proteins are phosphorylated by protein kinase A (PKA), casein kinase 1 (CK1) and glycogen synthase kinase-3β (GSK3β), which create binding sites for the E3 ubiquitin ligase β-transducing repeat-containing protein (β-TrCP). This promotes the complete proteasome-dependent degradation of GLI1. GLI2 and GLI3 are retained by SUFU in the cytoplasm [8][9][10][11], where they undergo partial proteasome degradation, leading to the formation of repressor forms (GLI3/2R) that translocate into the nucleus repressing the transcription of GLI target genes [12] (Figure 1). Degradation of β-TrCP by the endoplasmic reticulum aminopeptidase 1 (ERAP1), a key regulator of innate and adaptive immune responses [13], can protect GLI transcription factors from β-TrCP-dependent degradation and stimulate HH activity [14].
Canonical activation of HH signaling occurs upon the binding of the HH ligand to PTCH1, which exits the PC, relieving the inhibition of SMO and allowing the translocation of SMO into the PC [15]. Active SMO prevents GLI2 and GLI3 processing and promotes their dissociation from SUFU, leading to the translocation of full-length and active GLI (GLIACT) into the nucleus, where they activate the transcription of GLI target genes (Figure 1). Among them, there are GLI1 and PTCH1, which contribute to the creation of a positive feed-back loop. Other GLI targets include genes involved in cell proliferation (MYC, CCND1, CCND2, FOXM1), cell survival (BCL-2), angiogenesis (ANG1/2), epithelial-to-mesenchymal transition (SNAIL and ZEB), stemness (NANOG and SOX2) and several cytokines (IL-6, IL-1β and TNF-α) [16][17].
The HH signaling pathway is also activated through non-canonical mechanisms, which consist of the PTCH/SMO-dependent GLI-independent mechanism or in the activation of the GLI transcription factors independent of upstream PTCH/SMO. In the latter, the signal can bypass the canonical pathway to directly activate the GLI. This type of non-canonical activation occurs mainly in cancer cells and has been extensively investigated [18]. For instance, RAS-RAF-MEK-ERK1/2 and AKT signaling can regulate the nuclear localization and transcriptional activity of GLI1 in normal fibroblasts and melanoma cells [19][20][21]. In esophageal adenocarcinoma cells, the activation of mTOR signaling and S6K1 promotes the phosphorylation of GLI1 at Serine 84, preventing its association with SUFU [22]. Transforming growth factor β (TGFβ) is a strong inducer of both GLI1 and GLI2 in various human cell types, including normal fibroblasts and keratinocytes, as well as cancer cells [23]. Atypical protein kinase C ι/λ (aPKCι/λ) activates GLI1 through the phosphorylation of two residues (Ser243 and Thr304) in the zinc finger DNA binding domain of GLI1, leading to increased DNA binding and transcriptional activity [24]. The fusion oncogene Ewing Sarcoma/Friend Leukemia Integration 1 (EWS/FLI1) has been shown to induce GLI1 transcription via direct binding to the GLI1 promoter [25]. Aside from oncogenes, loss of tumor suppressors, such as p53 or the chromatin remodeling protein SNF5, have been shown to enhance the activity of GLI1 [26][27]. Furthermore, the epigenetic modulator bromodomain-containing protein 4 (BRD4) positively regulates HH signaling by directly binding to GLI1 and GLI2 promoters [28], and the histone deacetylase HDAC1 (Histone deacetylase 1) can deacetylate GLI1 at Lysine 518 to promote transcriptional activation [29].
HH signaling plays a critical role in several hallmarks of cancer, such as the sustaining of proliferative signals, evasion of growth suppression and cell death and activation of invasion and metastasis, inducing angiogenesis and immune evasion [2]. Uncontrolled activation of the HH pathway is involved in a variety of cancer types. HH signaling is a key driver in the pathogenesis of basal cell carcinoma (BCC), medulloblastoma (MB) and rhabdomyosarcoma. Moreover, aberrant activation of HH signaling has been implicated in the progression of gastrointestinal, pancreatic, liver, biliary tract, ovarian, breast, prostate and lung cancers, glioblastoma, melanoma and a number of hematological malignancies [30][31].
In light of the above, a great effort has been made in the last decade to develop inhibitors targeting the HH pathway. Current inhibitors against HH signaling include SMO and GLI antagonists. These molecules have been extensively reviewed elsewhere [32][33][34][35][36][37]; hence, only the most important among them will be mentioned.
Vismodegib (GDC-0449) was the first SMO inhibitor (SMOi) to be approved in 2012 for the treatment of locally advanced and metastatic BCC [38][39][40], followed in 2015 by sonidegib (LDE225), a potent and selective SMOi with high tissue penetration and the ability to cross the blood–brain barrier [41]. In 2018 the SMOi glasdegib (PF-04449913) was approved in combination with chemotherapy for the treatment of acute myeloid leukemia patients [42]. Other SMOi are in active clinical trials, including saridegib (IPI-926) [43] and taladegib (LY-2940680), which has shown efficacy in tumors harboring the SMO-D473H mutation, which causes drug resistance to vismodegib [44].
Despite the therapeutic efficacy of SMOi, the enthusiasm for their clinical use has been hampered by the development of primary or acquired resistance, and relapse upon drug withdrawal. Notably, about 50% of BCC patients developing resistance to SMOi present mutations in SMO, which occur in the drug-binding pocket of SMO or in other critical domains of the transmembrane helices [45][46]. Further resistance mechanisms include GLI2 gene amplification, and loss of the negative regulator SUFU. Inhibition of the GLI transcription factors represents an alternative strategy for the development of HH pathway inhibitors. This could be an effective approach against tumors resistant to SMOi and might have the dual advantage of blocking both the canonical and non-canonical HH pathway. To date, only a few GLI antagonists have been discovered and, except for arsenic trioxide (ATO), which is not a specific GLI inhibitor, their use has been limited to preclinical studies [33]. For instance, GANT61 and GANT58 have been shown to interfere with the binding of GLI to DNA and have shown efficacy in blocking tumor cell growth in vitro and in vivo [47]. The natural compound Glabrescione B also interferes with the interaction of GLI1/DNA and has shown therapeutic efficacy in preclinical models of HH-dependent cancers [48]. ATO, an already FDA-approved therapeutic for acute promyelocytic leukemia, has been found to suppress GLI1 transcriptional activity and block HH-induced ciliary accumulation of GLI2 [49][50]. ATO is currently in several clinical trials for cancer treatment as a single agent or in combinatorial regimen. More recently, a pharmacophore-based virtual screening approach identified quinolines and oxazino-quinoline derivatives as small molecule GLI1 inhibitors characterized by submicromolar antiproliferative activity toward human melanoma and medulloblastoma cell lines [51][52]. Further studies are in progress to optimize these small molecules and to assess their efficacy for the treatment of different types of cancer resistant to SMOi.
References
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087.
- Hanna, A.; Shevde, L.A. Hedgehog signaling: Modulation of cancer properties and tumor microenvironment. Mol. Cancer 2016, 15, 24.
- Grund-Gröschke, S.; Stockmaier, G.; Aberger, F. Hedgehog/GLI signaling in tumor immunity—New therapeutic opportunities and clinical implications. Cell Commun. Signal. 2019, 17, 172.
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472.
- Montagnani, V.; Stecca, B. Role of Protein Kinases in Hedgehog Pathway Control and Implications for Cancer Therapy. Cancers 2019, 11, 449.
- Svärd, J.; Heby-Henricson, K.; Persson-Lek, M.; Rozell, B.; Lauth, M.; Bergström, A.; Ericson, J.; Toftgård, R.; Teglund, S. Genetic elimination of suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell 2006, 10, 187–197.
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344.
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P.G. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1999, 1, 312–319.
- Wang, B.; Li, Y. Evidence for the direct involvement of βTrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 2006, 103, 33–38.
- Pan, Y.; Wang, B. A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J. Biol. Chem. 2007, 282, 10846–10852.
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181.
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377.
- Pepelyayeva, Y.; Amalfitano, A. The role of ERAP1 in autoinflammation and autoimmunity. Hum. Immunol. 2019, 80, 302–309.
- Bufalieri, F.; Infante, P.; Bernardi, F.; Caimano, M.; Romania, P.; Moretti, M.; Lospinoso Severini, L.; Talbot, J.; Melaiu, O.; Tanori, M.; et al. ERAP1 promotes Hedgehog-dependent tumorigenesis by controlling USP47-mediated degradation of βTrCP. Nat. Commun. 2019, 10, 3304.
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376.
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886.
- Sigafoos, A.N.; Paradise, B.D.; Fernandez-Zapico, M.E. Hedgehog/GLI Signaling Pathway: Transduction, Regulation, and Implications for Disease. Cancers 2021, 13, 3410.
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-Canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556.
- Riobo, N.A.; Haines, G.M.; Emerson, C.P., Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006, 66, 839–845.
- Riobó, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P., Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510.
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900.
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387.
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986.
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488.
- Zwerner, J.P.; Joo, J.; Warner, K.L.; Christensen, L.; Hu-Lieskovan, S.; Triche, T.J.; May, W.A. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2008, 27, 3282–3291.
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676.
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.; Tolstorukov, M.; et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433.
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740.
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142.
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812.
- McMillan, R.; Matsui, W. Molecular pathways: The hedgehog signaling pathway in cancer. Clin. Cancer Res. 2012, 18, 4883–4888.
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422.
- Infante, P.; Alfonsi, R.; Botta, B.; Mori, M.; Di Marcotullio, L. Targeting GLI factors to inhibit the Hedgehog pathway. Trends Pharm. Sci. 2015, 36, 547–558.
- Pietrobono, S.; Stecca, B. Targeting the Oncoprotein Smoothened by Small Molecules: Focus on Novel Acylguanidine Derivatives as Potent Smoothened Inhibitors. Cells 2018, 7, 272.
- Galperin, I.; Dempwolff, L.; Diederich, W.E.; Lauth, M. Inhibiting Hedgehog: An Update on Pharmacological Compounds and Targeting Strategies. J. Med. Chem. 2019, 62, 8392–8411.
- Gampala, S.; Yang, J.Y. Hedgehog Pathway Inhibitors against Tumor Microenvironment. Cells 2021, 10, 3135.
- Nguyen, N.M.; Cho, J. Hedgehog Pathway Inhibitors as Targeted Cancer Therapy and Strategies to Overcome Drug Resistance. Int. J. Mol. Sci. 2022, 23, 1733.
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 2502–2511.
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172.
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179.
- Pan, S.; Wu, X.; Jiang, J.; Gao, W.; Wan, Y.; Cheng, D.; Han, D.; Liu, J.; Englund, N.P.; Wang, Y.; et al. Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist. ACS Med. Chem. Lett. 2010, 1, 130–134.
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; LaGreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med. Chem. Lett. 2011, 3, 106–111.
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418.
- Bender, M.H.; Hipskind, P.A.; Capen, A.R.; Cockman, M.; Credille, K.M.; Gao, H.; Bastian, J.A.; Clay, J.M.; Lobb, K.L.; Sall, D.J.; et al. Identification and characterization of a novel smoothened antagonist for the treatment of cancer with deregulated hedgehog signaling. Cancer Res. 2011, 71, 2819.
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353.
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341.
- Lauth, M.; Bergström, A.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460.
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217.
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160.
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437.
- Manetti, F.; Stecca, B.; Santini, R.; Maresca, L.; Giannini, G.; Taddei, M.; Petricci, E. Pharmacophore-Based Virtual Screening for Identification of Negative Modulators of GLI1 as Potential Anticancer Agents. ACS Med. Chem. Lett. 2020, 11, 832–838.
- Manetti, F.; Maresca, L.; Crivaro, E.; Pepe, S.; Cini, E.; Singh, S.; Governa, P.; Maramai, S.; Giannini, G.; Stecca, B.; et al. Quinolines and Oxazino-quinoline Derivatives as Small Molecule GLI1 Inhibitors Identified by Virtual Screening. ACS Med. Chem. Lett. 2022, 13, 1329–1336.
More
Information
Subjects:
Cell Biology; Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.3K
Revisions:
3 times
(View History)
Update Date:
18 Jan 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No