Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manoj K. Pandey | -- | 2440 | 2023-01-15 21:43:50 | | | |
| 2 | Camila Xu | Meta information modification | 2440 | 2023-01-16 03:29:00 | | | | |
| 3 | Camila Xu | Meta information modification | 2440 | 2023-01-16 03:29:23 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Al-Odat, O.S.; Guirguis, D.A.; Schmalbach, N.K.; Yao, G.; Budak-Alpdogan, T.; Jonnalagadda, S.C.; Pandey, M.K. Autophagy and Apoptotic Pathways in Multiple Myeloma. Encyclopedia. Available online: https://encyclopedia.pub/entry/40186 (accessed on 06 August 2026).
Al-Odat OS, Guirguis DA, Schmalbach NK, Yao G, Budak-Alpdogan T, Jonnalagadda SC, et al. Autophagy and Apoptotic Pathways in Multiple Myeloma. Encyclopedia. Available at: https://encyclopedia.pub/entry/40186. Accessed August 06, 2026.
Al-Odat, Omar S., Daniel A. Guirguis, Nicole K. Schmalbach, Gabriella Yao, Tulin Budak-Alpdogan, Subash C. Jonnalagadda, Manoj K. Pandey. "Autophagy and Apoptotic Pathways in Multiple Myeloma" Encyclopedia, https://encyclopedia.pub/entry/40186 (accessed August 06, 2026).
Al-Odat, O.S., Guirguis, D.A., Schmalbach, N.K., Yao, G., Budak-Alpdogan, T., Jonnalagadda, S.C., & Pandey, M.K. (2023, January 15). Autophagy and Apoptotic Pathways in Multiple Myeloma. In Encyclopedia. https://encyclopedia.pub/entry/40186
Al-Odat, Omar S., et al. "Autophagy and Apoptotic Pathways in Multiple Myeloma." Encyclopedia. Web. 15 January, 2023.
Copy Citation
Multiple myeloma (MM) is caused by aberrant plasma cells (PCs) in the bone marrow (BM), representing 1% of neoplastic diseases and 13% of hematological neoplasms. MM is a challenging cancer to diagnose and treat.
multiple myeloma (MM)
autophagy
apoptosis
1. Introduction
Lymphoma, leukemia, and myeloma are the three main classifications for cancers of the hematopoietic system. Multiple myeloma (MM) is caused by aberrant plasma cells (PCs) in the bone marrow (BM), representing 1% of neoplastic diseases and 13% of hematological neoplasms [1]. MM is a challenging cancer to diagnose and treat. Patients go through two early stages of the disease before acquiring aggressive myeloma. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM) are the precursor stages with no clinical symptoms defined (Figure 1). Depending on the stage of the disease, the number of abnormal PCs, levels of monoclonal immunoglobulin (Ig), and cytogenetic abnormalities begin to rise, and ultimately patients become symptomatic (hypercalcemia, anemia, renal insufficiency, bone lesions, and multiple infections), and referred as MM. The American Cancer Society estimates that by the year 2022, approximately 34,470 new cases of MM will be diagnosed and 12,640 deaths from MM in the United States will occur [2].
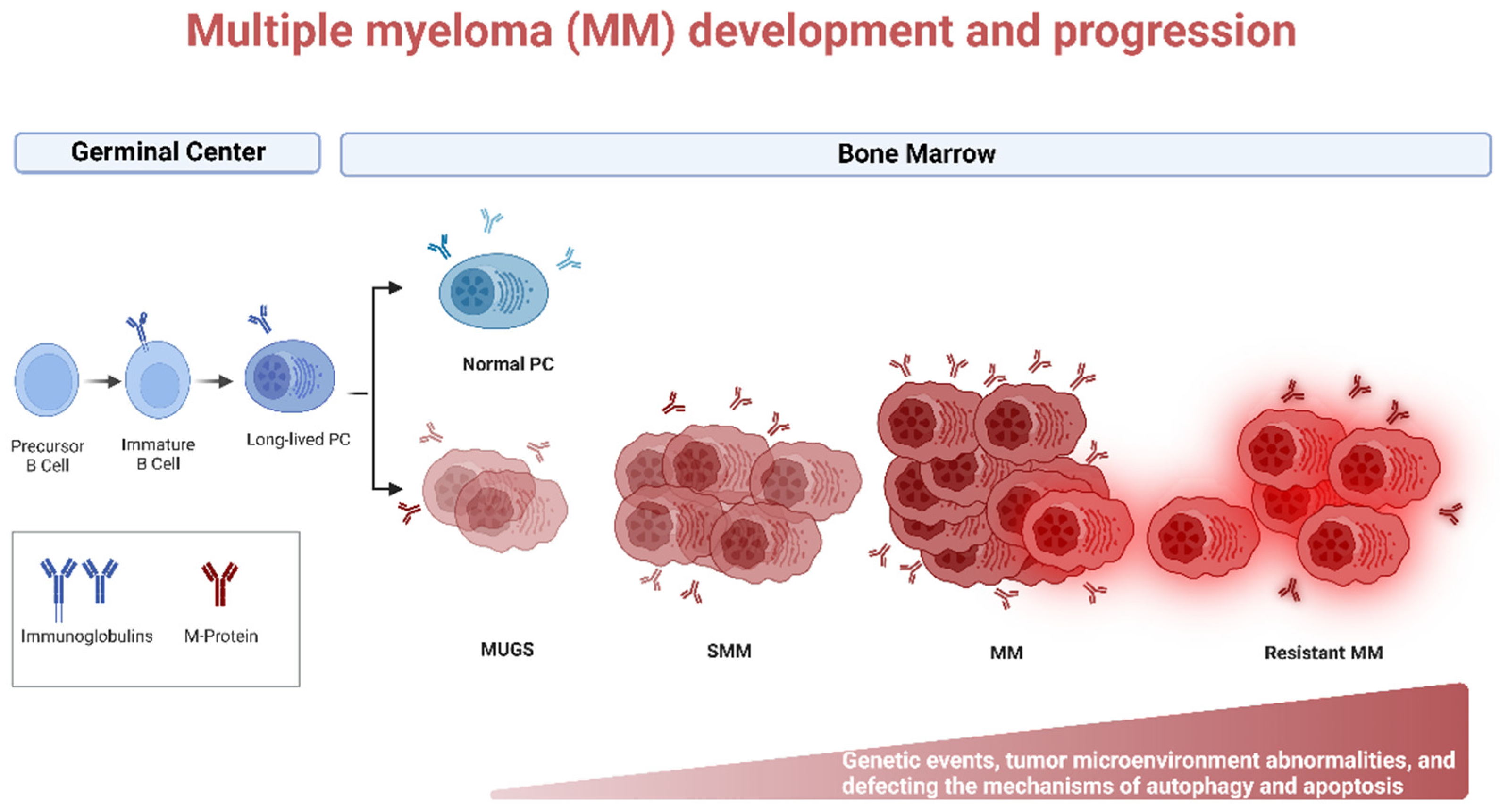
Figure 1. Biology of Multiple myeloma (MM) development and progression. MM is the end stage of a multistep neoplastic transformation of PCs. MGUS and SMM are the precursor stages of MM. Increase in aberrant PCs, Igs, and cytogenetic and tumor microenvironment abnormalities precede the onset of symptoms of MM. Relapsed/refractory (R/R) patients often present with a more aggressive phenotype. Genetic events, tumor microenvironment abnormalities, and defective autophagy and apoptosis may occur during MM progression. Created using Biorender.com (accessed on 1 November 2022).
The interaction of myeloma cells to bone marrow microenvironment (BMM) is the hallmark of MM [3]. Although treatment options for MM have improved remarkably over the last several decades, the survival rate remains extremely low, and all patients experience relapse or become refractory. Unsurprisingly, refractory and relapsed (R/R) patients often present with a more aggressive phenotype upon recurrence. It has been established that cytogenetic and epigenetic abnormalities play critical role in MM progression and resistance to available drugs. However, defective mechanisms of autophagy, apoptosis, tumor microenvironment, and cell survival signaling contribute in MM progression and drug resistance. Table 1 illustrates the common drugs that have been used to treat MM patients, its mechanism of action, and underlying mechanisms of drug resistance.
Table 1. Mechanism of action and underlying drug resistance mechanisms of the common chemotherapeutic agents.
| Class | Drugs | Mechanism of Action | Type of Resistance | Mechanism of Resistance |
|---|---|---|---|---|
| Alkalyting agents Anthracyclines |
Melphalan Cyclophosphamide Doxorubicin |
Impairment of DNA synthesis and cell replication, immunostimulatory activity by inhibiting interleukin-6 (IL6), interaction with dendritic cells, and immunogenic effects in the tumor microenvironment [4][5]. Topoisomerase II inhibition (Doxorubicin). | Alters autophagy and apoptosis signaling pathways Cancer stem cells and bone marrow microenvironment |
Upregulation of anti-apoptotic proteins (Mcl-1, Bcl-2, Bcl-xL) [6][7]. Stem cell-like phenotype with triggering of ALDH1A1 enzymatic activity and upregulation of BTK [8][9]. Increase of cell adhesion molecules (VLA4) [10]. |
| Proteasome inhibitors | Bortezomib Carfilzomib Ixazomib |
Inhibition of proteasome activity; inhibition of NF-κβ activity; induction of apoptosis by activation of caspase-8 and -9; upregulation of pro-apoptotic protein Noxa; downregulation of adhesion molecules on myeloma cells [4][11][12][13][14][15][16][17][18]. | Alters autophagy and apoptosis signaling pathways Cancer stem cells and bone marrow microenvironment |
Upregulation of the proteasomal system; Upregulation of anti-apoptotic proteins (Mcl-1, Bcl-2, Bcl-xL); activation of autophagy pathway; induction of NF-κβ; unfolded protein response (UPR) transcription factor XBP1 suppression; overexpression of heat shock proteins [3][19][20][21][22]. Stem cell-like phenotype with triggering of ALDH1A1 enzymatic activity and upregulation of BTK [8][9]. Secreting a group of extracellular signaling cues including IL-6, growth factors such as vascular endothelial growth factor (VEGF), and Insulin-like growth factor 1 (IGF-1); trigger and modulate multiple keys of the transcriptional pathway including Ras/MAPK, JAK/STAT3, and PI3/Akt; Increase of pro-inflammatory TNF-α; Increase of different cell adhesion molecules; overexpression of CXCR4; overexpression of MARCKS [22][23][24][25][26][27][28]. |
| Immunomodulatory agents | Thalidomide Lenalidomide Pomalidomide |
Induction of apoptosis by activation of cspase-8 and -9; interaction with BMM and downregulation of adhesion molecules; affecting cereblon (CRBN) and downstream targets; regulation of T and natural killer (NK) cells activity; anti-angiogenic activity [29][30]. | Cancer stem cells and bone marrow microenvironment | Stem cell-like phenotype with triggering of ALDH1A1 enzymatic activity and upregulation of BTK [8][9]. Downregulation of CRBN expression and deregulation of IRF4 expression; increased IL-6 expression and constitutive STAT3 activation [31]. |
| Histone deacetylase inhibitors | Panobinostat Vorinostat |
Increasing chromatin structure opening, end with activation of tumor suppressor genes [32][33][34]. | Bone marrow microenvironment and disruption intracellular signaling pathways | Regulation of actin cytoskeleton and protein processing in endoplasmic reticulum (triggering of MEK/ERK, PI3K, and FAK pathways) [35]. |
| Monoclonal antibodies | Daratumumab Elotuzumab Isatuximab |
Antibody-dependent cellular cytotoxicity (ADCC); complement-dependent cytotoxicity (CDC); modulation of target antigen enzymatic activity; macrophage-mediated pagocytosis; apoptosis via Fcγ receptor-mediated crosslinking; stimulation of immune cells activity, particularly T and NK cells [36][37][38][39][40][41]. | Bone marrow microenvironment | Competition by the soluble extracellular forms of CD38 and SLAM7 [42]. |
| Selective Exportin 1 (XPO1) inhibitor | Selinexor | Induces apoptosis through nuclear retention and functional activation of tumor suppressor proteins (TSPs), inhibits NF-κβ, and the translation of oncoprotein mRNAs [43][44][45]. | - | - |
| Corticosteroids | Dexamethasone Prednisolone Methylprednisolone |
Induction of apoptosis [46]. | Bone marrow microenvironment | Increased secretion of pro-survival cytokines bone marrow microenvironment [4][47]. |
2. Autoghagy in MM
Among the many essential biological functions of autophagy, it plays a critical role in innate and adaptive immunity. Autophagy helps eliminate various microorganisms, controls pro-inflammatory signaling and secretion of immune mediators, and controls homeostasis of lymphocyte and antigen presentation [48]. B lymphocytes include two distinct populations known as B1 and B2 cells, which vary distinctively in function and development. The B1 cells are involved in the innate immune system as opposed to the B2 cells, which are considered part of the adaptive immune system [49]. Autophagy is not necessary for B1 and B2 cell development but is required for B1 self-renewal in the periphery. Moreover, autophagy plays a vital role in the B2 cell’s Toll-like receptor (TLR) activation [50]. Pengo et al. demonstrated thatPCs have high autophagic activities and suggested that autophagy has a general role in PC oncogenesis. They also showed the viability of PCs as well as the importance of autophagy in sustaining antibody responses and its necessity in long-lived BM PCs [51]. Furthermore, Halliley et al. were able to demonstrate the importance of autophagy as a mechanism of survival in human long-lived PCs [52].
MM has similarities with the original plasma cell clone, such as an enlarged ER and cytoplasm, as well as immunoglobulin (Ig) secretion. MM cells produce a considerable amount of Ig, inevitably producing misfolded or unfolded proteins that can be toxic to the cells. MM uses various molecular pathways to protect the cells from damage and promote cell survival, including processes such as proteasomal degradation, UPR, and autophagy. In cases where there is excessive production of misfolded protein and the inability of the proteasomal system or UPR to deal with this stress, autophagy could be induced to help alleviate the stress. If all these survival mechanisms fail, apoptosis is engaged, and the cells get destroyed [53][54][55].
Autophagy in myeloma cells not only works in tandem with the ubiquitin-proteasome system (UPS) to remove ubiquitinated proteins [56], but it also plays a role in determining the susceptibility to proteasome inhibitors [57]. According to recent research on MM, greater immunoreactivity against autophagic markers including Beclin1 and LC3 corresponds with longer patient survival [58]. Furthermore, it has been shown that the imbalance between Beclin1 and p62 proteins expression promotes the proliferation of MM cells through autophagy regulation [59].
Aberrant DNA repair mechanisms contribute to illness development, initial translocations, and progression in MM. Melphalan (an alkylating agent) and bortezomib (a proteasome inhibitor) are two of the most common medications used to treat MM, they both work by inhibiting several steps in the DNA repair process that play a role in both the body’s response to therapy and any subsequent resistance [60]. DNA damage leads to the activation of autophagy, which plays a role in DNA damage response (DDR) and assists in cytokine secretion, senescence, cell death, and repairing DNA lesions [61]. DNA damage leads to the activation of ataxia-telangiectasia mutated (ATM) within the Mre11-Rad50-Nbs1 complex (MRN) that binds double-strand breaks. ATM is then able to activate AMPK that targets a TOR-Autophagy Spatial Coupling Compartment (TORC1) inhibitor known as tumor necrosis factor receptor (TNFR)-associated factor interacting protein 2 (TSC2). TORC1 acts as a negative regulator of the ULK complex; therefore, its inhibition allows autophagosome formation. ATM also regulates p53 and stabilizes it, and in turn, p53 regulates other autophagic pathways, including AMPK, PTEN, and damage-associated molecular pattern (DAPK). Furthermore, p53 modulates Sestrin, a protein that regulates TORC1 activity in an AMPK-independent manner through the GATOR2-GATOR1-RAGB/A (GTPase-activating protein activity toward RAGs) signaling pathway as well as via AMPK-dependent manner like how it affects TSC2. ATM activates Che-1, an RNA polymerase II-binding protein that increases the transcription of Deptor and Redd1 as well as inhibiting mTOR. ATM also modulates Beclin1 through the activation of the NF-ϰB essential modulator (NEMO)-dependent transforming growth factor (TAK1)-ATM-NEMO-NFϰB pathway. TAK1 can activate c-Jun N-terminal kinases (JNK), allowing Beclin1 to be released from antiapoptotic proteins. Autophagy also induced DNA damage by decreasing the transcription of MKP-1, a JNK phosphatase; thus, JNK induces autophagy. Moreover, DNA damage triggers an enzyme called PARP1, which in turn decreases cellular ATP and NAD+ levels, activating AMPK and initiating autophagy [61].
Cancer stem cells (CSC) could form cancers and self-renew, which is essential in resistance to anti-cancer agents. Autophagy is associated with the maintenance of CSC pluripotency and increases resistance to cancer drugs [62]. One of the prominent drugs used to treat MM patients is proteasome inhibitors (PIs). PIs cause an increase in the amount of misfolded or unfolded proteins, which increases ER stress. The cells adapt to this stress with the help of multiple pathways, including autophagy, which allows the cells to mitigate stressful environments and survive. Therefore, inducing autophagic cell death may potentially be an additional viable approach to dealing with MM. However, it has been shown that uncontrollable autophagy also promotes drug resistance in myeloma cells, and that blocking autophagy may restore sensitivity to medicines [63][64]. The most potent preclinical studies and clinical trials in MM involving autophagy inhibitor or inducer alone or in combination with bortezomib are summarized in Table 2.
Table 2. Preclinical studies and clinical trials of autophagy inhibitor or inducer alone or in combination with bortezomib in MM.
| Drug | Study Design | Clinical Trial Status |
|---|---|---|
| Autophagy inhibitors | ||
| Chloroquine (CQ) | In combination with bortezomib and cyclophosphamide in R/R MM patients | Phase II (NCT01438177) |
| Hydroxychloroquine (HCQ) | In combination with bortezomib and in R/R MM patients | Phase I/II (NCT00568880) |
| 3-Methyladenine (3-MA) | Human MM cell lines (U266, MM.1S, RPMI8226, and ARH 77) | Preclinical [65] |
| Bafilomycin A1 | In combination with bortezomib in U266 MM cell line | Preclinical [66] |
| Elaiophylin | Human MM cell lines (U266, RPMI8226, KMS11, and H929) | Preclinical [67] |
| Autophagy inducers | ||
| Metformin | Human MM cell lines (RPMI8226 and U266) and in vivo NOD-SCID murine xenograft MM model | Preclinical [68] |
3. Apoptotic Pathways in MM
Anti-apoptotic proteins play a major role in the pathogenesis of MM. MM cells exhibit imbalances in their anti-apoptotic protein’s expression levels, especially Mcl-1, that leads to prevent apoptosis and allow continued cell growth by inhibiting and forming heterodimer interaction with Bax and Bak proteins. Mcl-1 is known to be overexpressed in MM and plays a crucial role in MM initiation, progression, and chemoresistance [3]. Remarkably, 52% of newly diagnosed and 81% of relapsed MM patients have shown an increase in Mcl-1 protein expression, which correlates with disease progression and a poor patient survival rate [19]. Furthermore, the most common change in gene expression of MM is Mcl-1 overexpression. Approximately 40% of MM patients have increased expression of Mcl-1 and IL-6 receptors due to a gain or amplification in 1q21 [69]. Many studies currently are focused on advancing treatment therapies for MM and overcoming resistance challenges through inhibition of Mcl-1. Importantly, it has been established that Bcl-2 and Bcl-xL inhibitors such as venetoclax (ABT-199), ABT-737 and navitoclax (ABT-263), which mimics the BH3 domain of Bad and binds Bcl-2 and Bcl-xL, result in upregulation of Mcl-1 and develop resistance. Thus, inhibiting Mcl-1 represent a promising strategy in MM-sensitive and -resistance cells. Another promising avenue for targeting chemotherapeutic resistance via Mcl-1 is through Noxa. Proteasome inhibitors such as bortezomib increase expression of Noxa which selectively targets Mcl-1 for proteasomal degradation [70].
MM cells receive critical signals from the BMM that help them avoid apoptosis and preserve long-term survival (Figure 2). By secreting a set of signaling signals, BM stromal cells (BMSCs) control the expression of anti-apoptotic Bcl-2 family proteins particularly Mcl-1. Transcription of Mcl-1 is regulated via growth factors (e.g., VEGF, EGF), cytokines (e.g., IL-6, IL-5, IL-3), as well as cytotoxic stimuli. MM cells create an IL-6/VEGF loop to interact with BMM. Secretion of IL-6 from MM cells triggers IL-6 release in the BMSCs, inducing more VEGF secretion from the malignant cells and increasing proliferation [71]. In the BMM, MM plasma cells are activated by several factors such as IL-6, JAK/STAT, rat sarcoma/mitogen activated protein kinase (Ras/MAPK), phosphatidylinositol-3 kinase (PI3-K)/Akt, and TNF family including B cell activating factor (BAFF), and a proliferation inducing ligand (APRIL). Additionally, IGF-1 can play a role in the survival of MM cells, which can activate NF-κB and Akt, as well as increase expression of FLIP and cIAP-2 which inhibit caspase-8 [72]. IGF-1 can also downregulate the expression of Bim, resulting in less antagonism of anti-apoptotic proteins [73]. Upregulation of IGF-1 and IL-6 have also been associated as a phenotype of resistance in bortezomib resistant cells [74][75].
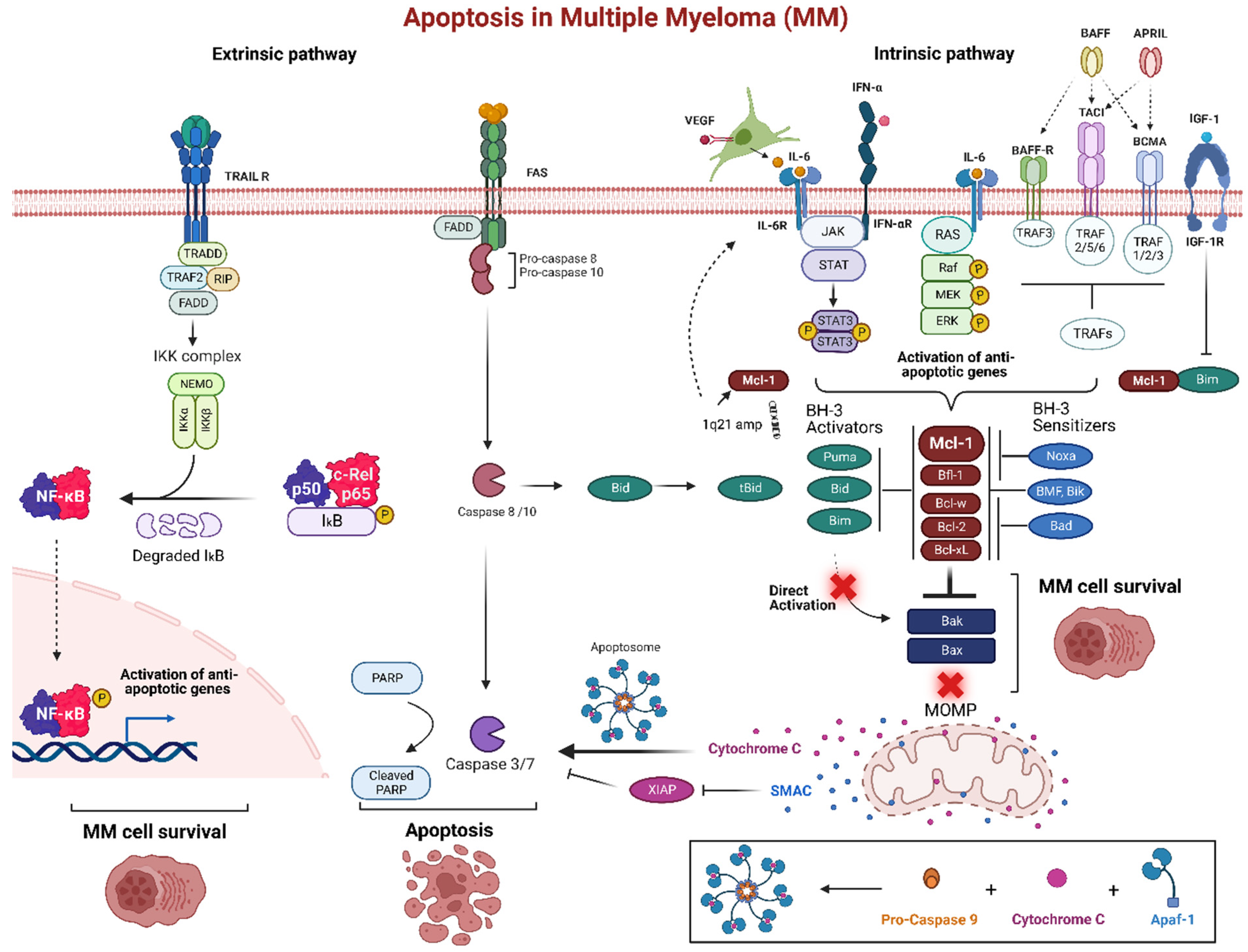
Figure 2. Apoptosis pathway in multiple myeloma (MM). The bone marrow microenvironment (BMM) is an important pathogenic factor for the long-term survival of MM. The intrinsic pathway of programmed cell death is promoted by signaling molecules from the BMM such as IL-6 and IFN-α that trigger various receptors in MM cells. These receptors, including tumor necrosis factor receptor-associated factors (TRAFs) through BAFF-R, BCMA, and TACI, as well as JAK/STAT or Ras/MAPK pathway activate anti-apoptotic proteins, particularly Mcl-1. Many MM patients develop increased expression of Mcl-1 and IL-6 via a gain or amplification of 1q21. This can create an IL-6/VEGF loop, as well as act as a mechanism of resistance through the upregulation of Mcl-1. Overexpression of anti-apoptotic proteins inhibits the BH3-only pro-apoptotic proteins and start competing for Bak and Bax binding site resulting MOMP inhibition. Normally, MOMP stimulation would release cytochrome c, forming the apoptosome after a conjunction with active caspase 9 and Apaf-1, to induce the executioner caspases 3 and 7 for apoptosis. The extrinsic pathway is triggered via the death receptors Fas and TRAIL. These receptors recruit FADD and pro-caspase-8 and 10 and can either directly activate caspase-8 and 10 to induce the downstream executioner caspases or activate Bid to induce MOMP. TRAIL receptor activation also activates anti-apoptotic genes via NF-κB complex. Created using Biorender.com (accessed on 12 December 2022).
References
- Palumbo, A.; Anderson, K. Venous thromboembolism in the patient with cancer: Focus on burden of disease and benefits of thromboprophylaxis. N. Engl. J. Med. 2011, 364, 1046–1060.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 71, 7–33.
- Al-Odat, O.; von Suskil, M.; Chitren, R.; Elbezanti, W.; Srivastava, S.; Budak-Alpddogan, T.; Jonnalagadda, S.; Aggarwal, B.; Pandey, M. Mcl-1 Inhibition: Managing Malignancy in Multiple Myeloma. Front. Pharmacol. 2021, 12, 699629.
- Pandey, M.; Amin, S.; Zangari, M.; Talamo, G. Drug resistance in multiple myeloma: How to cross the border. Ann. Hematol. Oncol. 2015, 2, 1025.
- Esma, F.; Salvini, M.; Troia, R.; Boccadoro, M.; Larocca, A.; Pautasso, C. Melphalan hydrochloride for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2017, 18, 1127–1136.
- Cho, H.J.; Mei, A.; Fukui, J.; Tung, K.; Leshchenko, V.V.; Lagana, A.; Patel, M.; Kim-Schulze, S.; Perumal, D.; Chari, A. MAGE-a mediate resistance to chemotherapy in multiple myeloma through regulation of Bcl-2 proteins. Blood 2016, 128, 3277.
- Tu, Y.; Renner, S.; Xu, F.-H.; Fleishman, A.; Taylor, J.; Weisz, J.; Vescio, R.; Rettig, M.; Berenson, J.; Krajewski, S. BCL-X expression in multiple myeloma: Possible indicator of chemoresistance. Cancer Res. 1998, 58, 256–262.
- Saltarella, I.; Lamanuzzi, A.; Reale, A.; Vacca, A.; Ria, R. Identify multiple myeloma stem cells: Utopia? World J. Stem Cells 2015, 7, 84.
- Franqui-Machin, R.; Wendlandt, E.B.; Janz, S.; Zhan, F.; Tricot, G. Cancer stem cells are the cause of drug resistance in multiple myeloma: Fact or fiction? Oncotarget 2015, 6, 40496.
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood J. Am. Soc. Hematol. 1999, 93, 1658–1667.
- Masaki, R. Mechanism of action of bortezomib in multiple myeloma therapy. Inte. J. Myeloma 2016, 6, 1–6.
- Mohan, M.; Matin, A.; Davies, F.E. Update on the optimal use of bortezomib in the treatment of multiple myeloma. Cancer Manag. Res. 2017, 9, 51.
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hájek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38.
- Franken, B.; van de Donk, N.; Cloos, J.; Zweegman, S.; Lokhorst, H. A clinical update on the role of carfilzomib in the treatment of relapsed or refractory multiple myeloma. Ther. Adv. Hematol. 2016, 7, 330–344.
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253.
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003, 348, 2609–2617.
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; Van Leeuwen, F.W.; Chanan-Khan, A.A. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood J. Am. Soc. Hematol. 2007, 110, 3281–3290.
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors–molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 2014, 18, 947–961.
- Wuilleme-Toumi, S.; Robillard, N.; Gomez, P.; Moreau, P.; Le Gouill, S.; Avet-Loiseau, H.; Harousseau, J.; Amiot, M.; Bataille, R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248–1252.
- Nikesitch, N.; Ling, S.C. Molecular mechanisms in multiple myeloma drug resistance. J. Clin. Pathol. 2016, 69, 97–101.
- Gambella, M.; Rocci, A.; Passera, R.; Gay, F.; Omedè, P.; Crippa, C.; Corradini, P.; Romano, A.; Rossi, D.; Ladetto, M. High XBP1 expression is a marker of better outcome in multiple myeloma patients treated with bortezomib. Haematologica 2014, 99, e14.
- Chatterjee, M.; Jain, S.; Stühmer, T.; Andrulis, M.; Ungethüm, U.; Kuban, R.J.; Lorentz, H.; Bommert, K.; Topp, M.; Krämer, D. STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90α and β in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood 2007, 109, 720–728.
- Hao, M.; Zhang, L.; An, G.; Sui, W.; Yu, Z.; Zou, D.; Xu, Y.; Chang, H.; Qiu, L. Suppressing miRNA-15a/-16 expression by interleukin-6 enhances drug-resistance in myeloma cells. J. Hematol. Oncol. 2011, 4, 37.
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.-T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E. BM mesenchymal stromal cell–derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555.
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910.
- Yang, Y.; Chen, Y.; Saha, M.; Chen, J.; Evans, K.; Qiu, L.; Reece, D.; Chen, G.A.; Chang, H. Targeting phospho-MARCKS overcomes drug-resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia 2015, 29, 715–726.
- Stessman, H.; Mansoor, A.; Zhan, F.; Janz, S.; Linden, M.; Baughn, L.; Van Ness, B. Reduced CXCR4 expression is associated with extramedullary disease in a mouse model of myeloma and predicts poor survival in multiple myeloma patients treated with bortezomib. Leukemia 2013, 27, 2075–2077.
- Stessman, H.A.; Baughn, L.B.; Sarver, A.; Xia, T.; Deshpande, R.; Mansoor, A.; Walsh, S.A.; Sunderland, J.J.; Dolloff, N.G.; Linden, M.A. Profiling bortezomib resistance identifies secondary therapies in a mouse myeloma model. Mol. Cancer Ther. 2013, 12, 1140–1150.
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2013, 54, 683–687.
- Hu, S.; Yuan, L.; Yan, H.; Li, Z. Design, synthesis and biological evaluation of Lenalidomide derivatives as tumor angiogenesis inhibitor. Bioorg. Med. Chem. Lett. 2017, 27, 4075–4081.
- Zhu, Y.X.; Shi, C.-X.; Bruins, L.A.; Wang, X.; Riggs, D.L.; Porter, B.; Ahmann, J.M.; de Campos, C.B.; Braggio, E.; Bergsagel, P.L. Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J. 2019, 9, 19.
- Afifi, S.; Michael, A.; Azimi, M.; Rodriguez, M.; Lendvai, N.; Landgren, O. Role of histone deacetylase inhibitors in relapsed refractory multiple myeloma: A focus on vorinostat and panobinostat. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2015, 35, 1173–1188.
- Yee, A.J.; Raje, N.S. Panobinostat and multiple myeloma in 2018. Oncologist 2018, 23, 516–517.
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the treatment of multiple myeloma. Clin. Cancer Res. 2015, 21, 4767–4773.
- Mithraprabhu, S.; Khong, T.; Spencer, A. Overcoming inherent resistance to histone deacetylase inhibitors in multiple myeloma cells by targeting pathways integral to the actin cytoskeleton. Cell Death Dis. 2014, 5, e1134.
- Varga, C.; Maglio, M.; Ghobrial, I.M.; Richardson, P.G. Current use of monoclonal antibodies in the treatment of multiple myeloma. Br. J. Haematol. 2018, 181, 447–459.
- De Weers, M.; Tai, Y.-T.; Van Der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848.
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood J. Am. Soc. Hematol. 2016, 128, 384–394.
- Overdijk, M.B.; Jansen, J.M.; Nederend, M.; van Bueren, J.J.L.; Groen, R.W.; Parren, P.W.; Leusen, J.H.; Boross, P. The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via Fcγ receptor–mediated cross-linking. J. Immunol. 2016, 197, 807–813.
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.; Briercheck, E.L. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849.
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; van Egmond, M.; van Bueren, J.J.L.; Mutis, T.; Groen, R.W.; Breij, E.; Martens, A.C.; Bleeker, W.K. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. In MAbs; Taylor & Francis: Abingdon, UK, 2015; pp. 311–320.
- van de Donk, N.W.; Moreau, P.; Plesner, T.; Palumbo, A.; Gay, F.; Laubach, J.P.; Malavasi, F.; Avet-Loiseau, H.; Mateos, M.-V.; Sonneveld, P. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood J. Am. Soc. Hematol. 2016, 127, 681–695.
- Chen, C.; Siegel, D.; Gutierrez, M.; Jacoby, M.; Hofmeister, C.C.; Gabrail, N.; Baz, R.; Mau-Sorensen, M.; Berdeja, J.G.; Savona, M. Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom macroglobulinemia. Blood J. Am. Soc. Hematol. 2018, 131, 855–863.
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S. Oral selinexor–dexamethasone for triple-class refractory multiple myeloma. N. Engl. J. Med. 2019, 381, 727–738.
- Vogl, D.T.; Dingli, D.; Cornell, R.F.; Huff, C.A.; Jagannath, S.; Bhutani, D.; Zonder, J.; Baz, R.; Nooka, A.; Richter, J. Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J. Clin. Oncol. 2018, 36, 859.
- Greenstein, S.; Ghias, K.; Krett, N.L.; Rosen, S.T. Mechanisms of glucocorticoid-mediated apoptosis in hematological malignancies. Clin. Cancer Res. 2002, 8, 1681–1694.
- Rees-Unwin, K.S.; Craven, R.A.; Davenport, E.; Hanrahan, S.; Totty, N.F.; Dring, A.M.; Banks, R.E.; Morgan, G.J.; Davies, F.E. Proteomic evaluation of pathways associated with dexamethasone-mediated apoptosis and resistance in multiple myeloma. Br. J. Haematol. 2007, 139, 559–567.
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737.
- Montecino-Rodriguez, E.; Dorshkind, K. New perspectives in B-1 B cell development and function. Trends Immunol. 2006, 27, 428–433.
- Raza, I.G.; Clarke, A.J. B cell metabolism and autophagy in autoimmunity. Front. Immunol. 2021, 2157.
- Pengo, N.; Scolari, M.; Oliva, L.; Milan, E.; Mainoldi, F.; Raimondi, A.; Fagioli, C.; Merlini, A.; Mariani, E.; Pasqualetto, E. Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol. 2013, 14, 298–305.
- Halliley, J.L.; Tipton, C.M.; Liesveld, J.; Rosenberg, A.F.; Darce, J.; Gregoretti, I.V.; Popova, L.; Kaminiski, D.; Fucile, C.F.; Albizua, I. Long-lived plasma cells are contained within the CD19− CD38hiCD138+ subset in human bone marrow. Immunity 2015, 43, 132–145.
- Benbrook, D.; Long, A. Integration of autography, proteasomal degradation, unfolded protein responce and apoptosis. Exp. Oncol. 2012, 3, 286–297.
- Hoang, B.; Benavides, A.; Shi, Y.; Frost, P.; Lichtenstein, A. Effect of autophagy on multiple myeloma cell viabilityAutophagy and Myeloma. Mol. Cancer Ther. 2009, 8, 1974–1984.
- Ho, M.; Patel, A.; Hanley, C.; Murphy, A.; McSweeney, T.; Zhang, L.; McCann, A.; O’Gorman, P.; Bianchi, G. Exploiting autophagy in multiple myeloma. J. Cancer Metastasis Treat. 2019, 5, 70.
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423.
- Milan, E.; Perini, T.; Resnati, M.; Orfanelli, U.; Oliva, L.; Raimondi, A.; Cascio, P.; Bachi, A.; Marcatti, M.; Ciceri, F. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015, 11, 1161–1178.
- Jung, G.; Roh, J.; Lee, H.; Gil, M.; Yoon, D.H.; Suh, C.; Jang, S.; Park, C.-J.; Huh, J.; Park, C.-S. Autophagic markers BECLIN1 and LC3 are associated with prognosis of multiple myeloma. Acta Haematol. 2015, 134, 17–24.
- Tucci, M.; Stucci, S.; Savonarola, A.; Resta, L.; Cives, M.; Rossi, R.; Silvestris, F. An imbalance between Beclin-1 and p62 expression promotes the proliferation of myeloma cells through autophagy regulation. Exp. Hematol. 2014, 42, 897–908.
- Gourzones-Dmitriev, C.; Kassambara, A.; Sahota, S.; Rème, T.; Moreaux, J.; Bourquard, P.; Hose, D.; Pasero, P.; Constantinou, A.; Klein, B. DNA repair pathways in human multiple myeloma: Role in oncogenesis and potential targets for treatment. Cell Cycle 2013, 12, 2760–2773.
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA damage response and autophagy: A meaningful partnership. Front. Genet. 2016, 7, 204.
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718.
- Malek, M.A.A.; Jagannathan, S.; Malek, E.; Sayed, D.M.; Elgammal, S.A.; Abd El-Azeem, H.G.; Thabet, N.M.; Driscoll, J.J. Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 2015, 6, 3098.
- Hamouda, M.-A.; Belhacene, N.; Puissant, A.; Colosetti, P.; Robert, G.; Jacquel, A.; Mari, B.; Auberger, P.; Luciano, F. The small heat shock protein B8 (HSPB8) confers resistance to bortezomib by promoting autophagic removal of misfolded proteins in multiple myeloma cells. Oncotarget 2014, 5, 6252.
- Shanmugam, M.; McBrayer, S.K.; Qian, J.; Raikoff, K.; Avram, M.J.; Singhal, S.; Gandhi, V.; Schumacker, P.T.; Krett, N.L.; Rosen, S.T. Targeting glucose consumption and autophagy in myeloma with the novel nucleoside analogue 8-aminoadenosine. J. Biol. Chem. 2009, 284, 26816–26830.
- Kawaguchi, T.; Miyazawa, K.; Moriya, S.; Ohtomo, T.; Che, X.-F.; Naito, M.; Itoh, M.; Tomoda, A. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: Crosstalk among proteasome, autophagy-lysosome and ER stress. Int. J. Oncol. 2011, 38, 643–654.
- Wang, G.; Zhou, P.; Chen, X.; Zhao, L.; Tan, J.; Yang, Y.; Fang, Y.; Zhou, J. The novel autophagy inhibitor elaiophylin exerts antitumor activity against multiple myeloma with mutant TP53 in part through endoplasmic reticulum stress-induced apoptosis. Cancer Biol. Ther. 2017, 18, 584–595.
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63.
- Slomp, A.; Moesbergen, L.M.; Gong, J.-N.; Cuenca, M.; von dem Borne, P.A.; Sonneveld, P.; Huang, D.C.; Minnema, M.C.; Peperzak, V. Multiple myeloma with 1q21 amplification is highly sensitive to MCL-1 targeting. Blood Adv. 2019, 3, 4202–4214.
- Podar, K.; Gouill, S.L.; Zhang, J.; Opferman, J.T.; Zorn, E.; Tai, Y.T.; Hideshima, T.; Amiot, M.; Chauhan, D.; Harousseau, J.L.; et al. A pivotal role for Mcl-1 in Bortezomib-induced apoptosis. Oncogene 2008, 27, 721–731.
- Dankbar, B.; Padró, T.; Leo, R.; Feldmann, B.; Kropff, M.; Mesters, R.M.; Serve, H.; Berdel, W.E.; Kienast, J. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood J. Am. Soc. Hematol. 2000, 95, 2630–2636.
- Mitsiades, C.S.; Mitsiades, N.; Poulaki, V.; Schlossman, R.; Akiyama, M.; Chauhan, D.; Hideshima, T.; Treon, S.P.; Munshi, N.C.; Richardson, P.G.; et al. Activation of NF-κB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: Therapeutic implications. Oncogene 2002, 21, 5673–5683.
- De Bruyne, E.; Bos, T.J.; Schuit, F.; Van Valckenborgh, E.; Menu, E.; Thorrez, L.; Atadja, P.; Jernberg-Wiklund, H.; Vanderkerken, K. IGF-1 suppresses Bim expression in multiple myeloma via epigenetic and posttranslational mechanisms. Blood J. Am. Soc. Hematol. 2010, 115, 2430–2440.
- Kuhn, D.J.; Berkova, Z.; Jones, R.J.; Woessner, R.; Bjorklund, C.C.; Ma, W.; Davis, R.E.; Lin, P.; Wang, H.; Madden, T.L.; et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 2012, 120, 3260–3270.
- Liu, Y.; Cheng, P.; Zhao, W.; Zhu, L.; Sui, J.; Dai, Y.; Lai, Y. MiR-197-3p reduces bortezomib resistance in multiple myeloma by inhibiting IL-6 expression in a MEAF6-dependent manner. Leuk. Res. 2022, 114, 106785.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.1K
Revisions:
3 times
(View History)
Update Date:
16 Jan 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No