 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | George Anderson | -- | 3825 | 2023-01-06 16:30:45 | | | |
| 2 | Jessie Wu | + 386 word(s) | 4211 | 2023-01-09 04:56:16 | | | | |
| 3 | Jessie Wu | -1 word(s) | 4210 | 2023-01-09 04:58:46 | | | | |
| 4 | Jessie Wu | -38 word(s) | 4172 | 2023-01-09 05:01:18 | | |
Video Upload Options
Amyotrophic lateral sclerosis (ALS) is a traumatic condition, whereby motor neurons are lost eventually leading to death within a couple of years. There is no effective treatment, due primarily to a lack of understanding as to the relevant pathophysiological changes occurring. The biological underpinnings of ALS is described, highlighting the role of the gut microbiome, glia, muscles and the changes at the neuromuscular junction. It is proposed that alterations in the mitochondrial melatonergic pathway are a crucial aspect of ALS pathophysiology that underpins the changes in intercellular interactions that drive ALS. Many of these initial changes seem to arise in the gut, indicating that this may be an important site for evaluation and treatment in the prevention of ALS. It is also proposed that glyphosate-based herbicides (weedkillers) exposure may be a relevant aspect of ALS susceptibility and progression.
1. Introduction
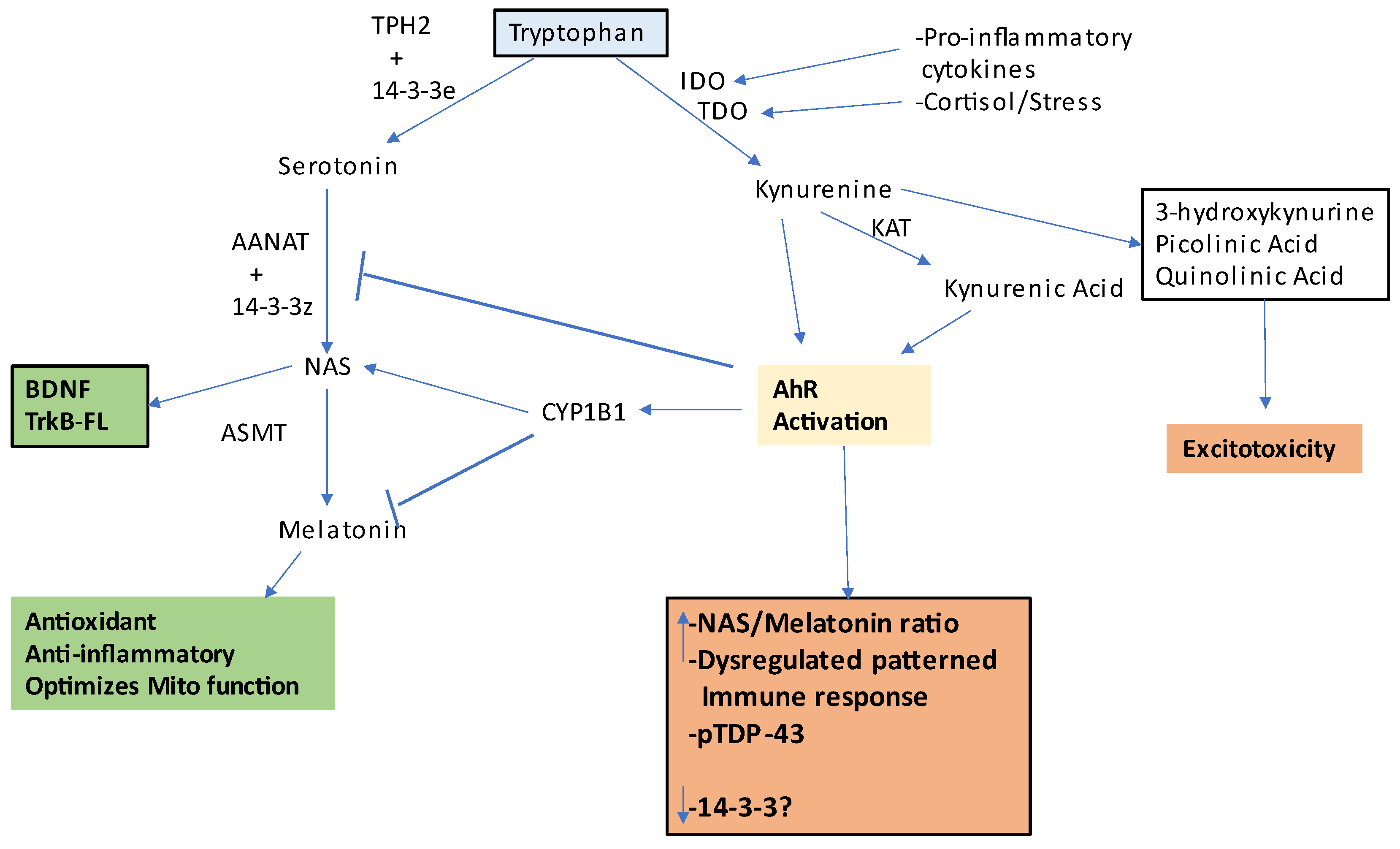
2. Integrating Amyotrophic Lateral Sclerosis Pathophysiology
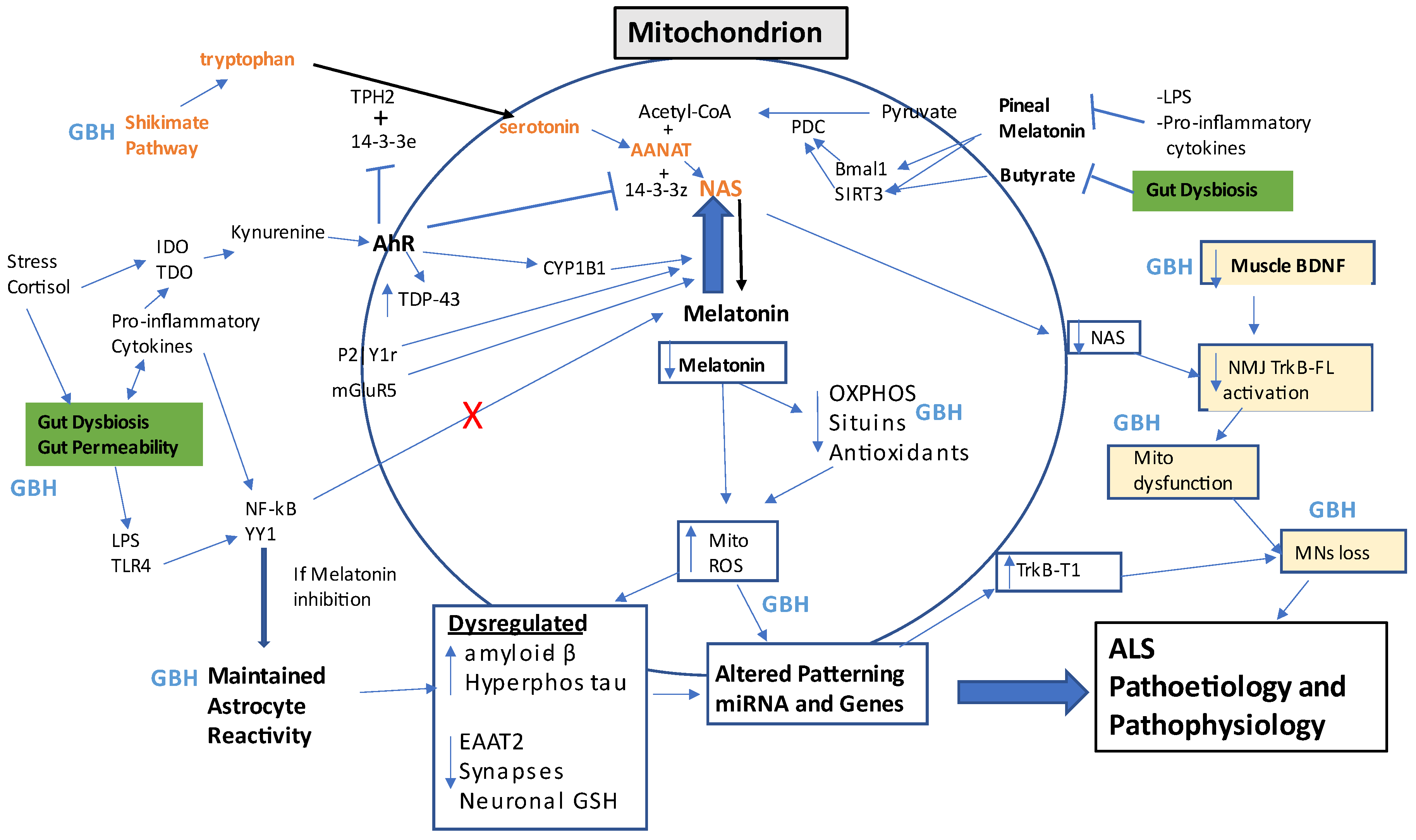
Glyphosate-Based Herbicides and Integrated Amyotrophic Lateral Sclerosis Pathophysiology
| GBH Effect | Glia | Gut | Muscles | NMJ | References |
|---|---|---|---|---|---|
| Shikimate pathway inhibition; |
Decrease tryptophan, 5-HT, NAS, melatonin? |
Altered gut bacteria patterning; |
Altered 5-HT muscle regulation? |
Altered MNs and NMJ 5-HT? | [74][75] |
| increased gut permeability, circulating LPS | Reactivation, Inflammation, Increased YY1, lower EAATs |
Altered gut bacteria interactions with mucosal immune system and enteric glia. |
Muscle NF-kB driven inflammation | MNs apoptotic processes | [76][77] |
| GBH resistant gut bacteria | Lower butyrate/HDACi-linked epigenetic regulation. Altered fungal composition |
Gut dysbiosis, HDACi | Lower butyrate HDACi-linked epigenetic regulation. Mitochondrial dysfunction. |
Lower butyrate, HDACi-linked epigenetic regulation. |
[76][78] |
| Increased TNF-α and amyloid-β |
Reactivation, Inflammation, |
Gut permeability |
Muscle damage Lower muscle BDNF | MNs loss | [62][79][80][81][82][83] |
| Increase miR-34a, other miRNAs? | Lower TrkB-FL/TrkBT1 ratio | Gut permeability |
Suboptimal muscle function |
Lower TrkB-FL/TrkBT1 ratio | [62][83][84] |
| Suppresses α7nAChR | Lost attenuation of glia and immune reactivity | Gut permeability |
Heightened muscle inflammation |
Heightened MNs and NMJ inflammation |
[85][86][87] |
| Platelet dysregulation |
BBB permeability | Gut permeability |
Muscle atrophy | ? | [14][67][68][69][70][71][72] |
References
- Edgar, S.; Ellis, M.; Abdul-Aziz, N.A.; Goh, K.J.; Shahrizaila, N.; Kennerson, M.L.; Ahmad-Annuar, A. Mutation analysis of SOD1, C9orf72, TARDBP and FUS genes in ethnically-diverse Malaysian patients with amyotrophic lateral sclerosis (ALS). Neurobiol. Aging 2021, 108, 200–206.
- Gurfinkel, Y.; Polain, N.; Sonar, K.; Nice, P.; Mancera, R.L.; Rea, S.L. Functional and structural consequences of TBK1 missense variants in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurobiol. Dis. 2022, 174, 105859.
- Rayner, S.L.; Hogan, A.; Davidson, J.M.; Cheng, F.; Lou, L.; Morsch, M.; Blair, I.; Chung, R.; Lee, A. Cyclin F, Neurodegeneration, and the Pathogenesis of ALS/FTD. Neuroscientist 2022.
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007, 10, 615–622.
- Wang, M.J.; Kang, L.; Wang, Y.Z.; Yang, B.R.; Zhang, C.; Lu, Y.F.; Kang, L. Microglia in motor neuron disease: Signaling evidence from last 10 years. Dev. Neurobiol. 2022, 82, 625–638.
- Almad, A.A.; Taga, A.; Joseph, J.; Gross, S.K.; Welsh, C.; Patankar, A.; Richard, J.P.; Rust, K.; Pokharel, A.; Plott, C.; et al. Cx43 hemichannels contribute to astrocyte-mediated toxicity in sporadic and familial ALS. Proc. Natl. Acad. Sci. USA 2022, 119, e2107391119.
- Gong, Z.; Ba, L.; Zhang, M. Dysfunction of the oligodendrocytes in amyotrophic lateral sclerosis. J. Biomed. Res. 2022, 36, 336–342.
- Badu-Mensah, A.; Guo, X.; Nimbalkar, S.; Cai, Y.; Hickman, J.J. ALS mutations in both human skeletal muscle and motoneurons differentially affects neuromuscular junction integrity and function. Biomaterials 2022, 289, 121752.
- Harrison, J.M.; Rafuse, V.F. Muscle fiber-type specific terminal Schwann cell pathology leads to sprouting deficits following partial denervation in SOD1G93A mice. Neurobiol. Dis. 2020, 145, 105052.
- Verma, S.; Khurana, S.; Vats, A.; Sahu, B.; Ganguly, N.K.; Chakraborti, P.; Gourie-Devi, M.; Taneja, V. Neuromuscular Junction Dysfunction in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2022, 59, 1502–1527.
- Martin, S.; Battistini, C.; Sun, J. A Gut Feeling in Amyotrophic Lateral Sclerosis: Microbiome of Mice and Men. Front Cell Infect Microbiol. 2022, 12, 839526.
- Pattle, S.B.; O’Shaughnessy, J.; Kantelberg, O.; Rifai, O.M.; Pate, J.; Nellany, K.; Hays, N.; Arends, M.J.; Horrocks, M.H.; Waldron, F.M.; et al. pTDP-43 aggregates accumulate in non-central nervous system tissues prior to symptom onset in amyotrophic lateral sclerosis: A case series linking archival surgical biopsies with clinical phenotypic data. J. Pathol. Clin. Res. 2023, 9, 44–55.
- Anderson, G.; Maes, M. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis via Suboptimal Mitochondrial Function: Assessment, Treatment and Classification Implications. Curr. Top. Med. Chem. 2020, 20, 524–539.
- Anderson, G.; Rodriguez, M.; Reiter, R.J. Multiple Sclerosis: Melatonin, Orexin, and Ceramide Interact with Platelet Activation Coagulation Factors and Gut-Microbiome-Derived Butyrate in the Circadian Dysregulation of Mitochondria in Glia and Immune Cells. Int. J. Mol. Sci. 2019, 20, 5500.
- Kaur, K.; Chen, P.C.; Ko, M.W.; Mei, A.; Chovatiya, N.; Huerta-Yepez, S.; Ni, W.; Mackay, S.; Zhou, J.; Maharaj, D.; et al. The Potential Role of Cytotoxic Immune Effectors in Induction, Progression and Pathogenesis of Amyotrophic Lateral Sclerosis (ALS). Cells 2022, 11, 3431.
- Appel, S.H.; Beers, D.R.; Zhao, W. Amyotrophic lateral sclerosis is a systemic disease: Peripheral contributions to inflammation-mediated neurodegeneration. Curr. Opin Neurol. 2021, 34, 765–772.
- Yanpallewar, S.; Fulgenzi, G.; Tomassoni-Ardori, F.; Barrick, C.; Tessarollo, L. Delayed onset of inherited ALS by deletion of the BDNF receptor TrkB.T1 is non-cell autonomous. Exp. Neurol. 2021, 337, 113576.
- Choi, B.J.; Park, K.H.; Park, M.H.; Huang, E.J.; Kim, S.H.; Bae, J.S.; Jin, H.K. Acid sphingomyelinase inhibition improves motor behavioral deficits and neuronal loss in an amyotrophic lateral sclerosis mouse model. BMB Rep. 2022, 55, 621–626.
- McCluskey, G.; Donaghy, C.; Morrison, K.E.; McConville, J.; Duddy, W.; Duguez, S. The Role of Sphingomyelin and Ceramide in Motor Neuron Diseases. J. Pers. Med. 2022, 12, 1418.
- Ash, P.E.A.; Stanford, E.A.; Al Abdulatif, A.; Ramirez-Cardenas, A.; Balance, H.I.; Boudeau, S.; Jeh, A.; Murithi, J.M.; Tripodis, Y.; Murphy, G.J.; et al. Dioxins and related environmental contaminants increase TDP-43 levels. Mol. Neurodegener. 2017, 12, 35.
- Alarcan, H.; Chaumond, R.; Emond, P.; Benz-De Bretagne, I.; Lefèvre, A.; Bakkouche, S.E.; Veyrat-Durebex, C.; Vourc’h, P.; Andres, C.; Corcia, P.; et al. Some CSF Kynurenine Pathway Intermediates Associated with Disease Evolution in Amyotrophic Lateral Sclerosis. Biomolecules 2021, 11, 691.
- Fifita, J.A.; Chan Moi Fat, S.; McCann, E.P.; Williams, K.L.; Twine, N.A.; Bauer, D.C.; Rowe, D.B.; Pamphlett, R.; Kiernan, M.C.; Tan, V.X.; et al. Genetic Analysis of Tryptophan Metabolism Genes in Sporadic Amyotrophic Lateral Sclerosis. Front. Immunol. 2021, 12, 701550.
- Bald, E.M.; Nance, C.S.; Schultz, J.L. Melatonin may slow disease progression in amyotrophic lateral sclerosis: Findings from the Pooled Resource Open-Access ALS Clinic Trials database. Muscle Nerve 2021, 63, 572–576.
- Huang, Z.; Liu, Q.; Peng, Y.; Dai, J.; Xie, Y.; Chen, W.; Long, S.; Pei, Z.; Su, H.; Yao, X. Circadian Rhythm Dysfunction Accelerates Disease Progression in a Mouse Model With Amyotrophic Lateral Sclerosis. Front. Neurol. 2018, 9, 218.
- Zhang, T.; Jiang, X.; Xu, M.; Wang, H.; Sang, X.; Qin, M.; Bao, P.; Wang, R.; Zhang, C.; Lu, H.; et al. Sleep and circadian abnormalities precede cognitive deficits in R521C FUS knockin rats. Neurobiol. Aging 2018, 72, 159–170.
- Ju, Y.E.; McLeland, J.S.; Toedebusch, C.D.; Xiong, C.; Fagan, A.M.; Duntley, S.P.; Morris, J.C.; Holtzman, D.M. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013, 70, 587–593.
- Anderson, G.; Maes, M. Interactions of Tryptophan and Its Catabolites With Melatonin and the Alpha 7 Nicotinic Receptor in Central Nervous System and Psychiatric Disorders: Role of the Aryl Hydrocarbon Receptor and Direct Mitochondria Regulation. Int. J. Tryptophan Res. 2017, 10, 1178646917691738.
- Anderson, G.; Maes, M. The Gut-Brain Axis: The Role of Melatonin in Linking Psychiatric, Inflammatory and Neurodegenerative Conditions. Adv. Integr. Med. 2015, 2, 31–37.
- Saitoh, Y.; Takahashi, Y. Riluzole for the treatment of amyotrophic lateral sclerosis. Neurodegener. Dis. Manag. 2020, 10, 343–355.
- Ortiz, J.F.; Khan, S.A.; Salem, A.; Lin, Z.; Iqbal, Z.; Jahan, N. Post-Marketing Experience of Edaravone in Amyotrophic Lateral Sclerosis: A Clinical Perspective and Comparison With the Clinical Trials of the Drug. Cureus 2020, 12, e10818.
- He, P.; He, B.; Li, S.; Chai, W.; Rao, W.; Zhu, Y.; Chen, W.; Zhang, P.; Zhang, X.; Pan, H.; et al. Distribution Features and Potential Effects of Serotonin in the Cerebrum of SOD1 G93A Transgenic Mice. eNeuro. 2022, 9, ENEURO.0001-22.2022.
- Dupuis, L.; Spreux-Varoquaux, O.; Bensimon, G.; Jullien, P.; Lacomblez, L.; Salachas, F.; Bruneteau, G.; Pradat, P.F.; Loeffler, J.P.; Meininger, V. Platelet serotonin level predicts survival in amyotrophic lateral sclerosis. PLoS ONE 2010, 5, e13346.
- Fiscon, G.; Conte, F.; Amadio, S.; Volonté, C.; Paci, P. Drug Repurposing: A Network-based Approach to Amyotrophic Lateral Sclerosis. Neurotherapeutics 2021, 18, 1678–1691.
- Novak, V.; Rogelj, B.; Župunski, V. Therapeutic Potential of Polyphenols in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Antioxidants 2021, 10, 1328.
- Yun, Y.C.; Jeong, S.G.; Kim, S.H.; Cho, G.W. Reduced sirtuin 1/adenosine monophosphate-activated protein kinase in amyotrophic lateral sclerosis patient-derived mesenchymal stem cells can be restored by resveratrol. J. Tissue Eng. Regen. Med. 2019, 13, 110–115.
- Ip, P.; Sharda, P.R.; Cunningham, A.; Chakrabartty, S.; Pande, V.; Chakrabartty, A. Quercitrin and quercetin 3-β-d-glucoside as chemical chaperones for the A4V SOD1 ALS-causing mutant. Protein Eng. Des. Sel. 2017, 30, 431–440.
- Sun, Y.; Bedlack, R.; Armon, C.; Beauchamp, M.; Bertorini, T.; Bowser, R.; Bromberg, M.; Caress, J.; Carter, G.; Crayle, J.; et al. ALSUntangled #64: Butyrates. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2022, 23, 638–643.
- Klingl, Y.E.; Pakravan, D.; Van Den Bosch, L. Opportunities for histone deacetylase inhibition in amyotrophic lateral sclerosis. Br. J. Pharmacol. 2021, 178, 1353–1372.
- Parrella, E.; Porrini, V.; Scambi, I.; Gennari, M.M.; Gussago, C.; Bankole, O.; Benarese, M.; Mariotti, R.; Pizzi, M. Synergistic association of resveratrol and histone deacetylase inhibitors as treatment in amyotrophic lateral sclerosis. Front. Pharmacol. 2022, 13, 1017364.
- Anderson, G. Tumour Microenvironment: Roles of the Aryl Hydrocarbon Receptor, O-GlcNAcylation, Acetyl-CoA and Melatonergic Pathway in Regulating Dynamic Metabolic Interactions across Cell Types-Tumour Microenvironment and Metabolism. Int. J. Mol. Sci. 2020, 22, 141.
- Seyfried, T.N.; Chinopoulos, C. Can the Mitochondrial Metabolic Theory Explain Better the Origin and Management of Cancer than Can the Somatic Mutation Theory? Metabolites 2021, 11, 572.
- Gevezova, M.; Sarafian, V.; Anderson, G.; Maes, M. Inflammation and Mitochondrial Dysfunction in Autism Spectrum Disorder. CNS Neurol. Disord Drug Targets 2020, 19, 320–333.
- Anderson, G.; Maes, M. Mitochondria and immunity in chronic fatigue syndrome. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 103, 109976.
- Mazzoccoli, G.; Kvetnoy, I.; Mironova, E.; Yablonskiy, P.; Sokolovich, E.; Krylova, J.; Carbone, A.; Anderson, G.; Polyakova, V. The melatonergic pathway and its interactions in modulating respiratory system disorders. Biomed. Pharmacother. 2021, 137, 111397.
- Brasanac, J.; Gamradt, S.; Otte, C.; Milaneschi, Y.; Monzel, A.S.; Picard, M.; Gold, S.M. Cellular specificity of mitochondrial and immunometabolic features in major depression. Mol. Psychiatry 2022, 27, 2370–2371.
- Acuña-Castroviejo, D.; Rahim, I.; Acuña-Fernández, C.; Fernández-Ortiz, M.; Solera-Marín, J.; Sayed, R.K.A.; Díaz-Casado, M.E.; Rusanova, I.; López, L.C.; Escames, G. Melatonin, clock genes and mitochondria in sepsis. Cell Mol. Life Sci. 2017, 74, 3965–3987.
- Zhao, X.; Shu, F.; Wang, X.; Wang, F.; Wu, L.; Li, L.; Lv, H. Inhibition of microRNA-375 ameliorated ketamine-induced neurotoxicity in human embryonic stem cell derived neurons. Eur. J. Pharmacol. 2019, 844, 56–64.
- Li, B.; Jiang, Y.; Xu, Y.; Li, Y.; Li, B. Identification of miRNA-7 as a regulator of brain-derived neurotrophic factor/α-synuclein axis in atrazine-induced Parkinson’s disease by peripheral blood and brain microRNA profiling. Chemosphere 2019, 233, 542–548.
- Lin, D.T.; Wu, J.; Holstein, D.; Upadhyay, G.; Rourk, W.; Muller, E.; Lechleiter, J.D. Ca2+ signaling, mitochondria and sensitivity to oxidative stress in aging astrocytes. Neurobiol. Aging 2007, 28, 99–111.
- Markus, R.P.; Fernandes, P.A.; Kinker, G.S.; da Silveira Cruz-Machado, S.; Marçola, M. Immune-pineal axis—Acute inflammatory responses coordinate melatonin synthesis by pinealocytes and phagocytes. Br. J. Pharmacol. 2018, 175, 3239–3250.
- Muxel, S.M.; Pires-Lapa, M.A.; Monteiro, A.W.; Cecon, E.; Tamura, E.K.; Floeter-Winter, L.M.; Markus, R.P. NF-κB drives the synthesis of melatonin in RAW 264.7 macrophages by inducing the transcription of the arylalkylamine-N-acetyltransferase (AA-NAT) gene. PLoS ONE 2012, 7, e52010.
- Rahman, M.H.; Bhusal, A.; Kim, J.H.; Jha, M.K.; Song, G.J.; Go, Y.; Jang, I.S.; Lee, I.K.; Suk, K. Astrocytic pyruvate dehydrogenase kinase-2 is involved in hypothalamic inflammation in mouse models of diabetes. Nat. Commun. 2020, 11, 5906.
- Pukhalskaia, A.E.; Dyatlova, A.S.; Linkova, N.S.; Kozlov, K.L.; Kvetnaia, T.V.; Koroleva, M.V.; Kvetnoy, I.M. Sirtuins as Possible Predictors of Aging and Alzheimer’s Disease Development: Verification in the Hippocampus and Saliva. Bull. Exp. Biol. Med. 2020, 169, 821–824.
- Jha, M.K.; Song, G.J.; Lee, M.G.; Jeoung, N.H.; Go, Y.; Harris, R.A.; Park, D.H.; Kook, H.; Lee, I.K.; Suk, K. Metabolic Connection of Inflammatory Pain: Pivotal Role of a Pyruvate Dehydrogenase Kinase-Pyruvate Dehydrogenase-Lactic Acid Axis. J. Neurosci. 2015, 35, 14353–14369.
- Salminen, A. Aryl hydrocarbon receptor (AhR) reveals evidence of antagonistic pleiotropy in the regulation of the aging process. Cell Mol. Life Sci. 2022, 79, 489.
- Mokrane, N.; Snabi, Y.; Cens, T.; Guiramand, J.; Charnet, P.; Bertaud, A.; Menard, C.; Rousset, M.; de Jesus Ferreira, M.C.; Thibaud, J.B.; et al. Manipulations of Glutathione Metabolism Modulate IP3-Mediated Store-Operated Ca2+ Entry on Astroglioma Cell Line. Front. Aging Neurosci. 2021, 13, 785727.
- Lee, M.; Cho, T.; Jantaratnotai, N.; Wang, Y.T.; McGeer, E.; McGeer, P.L. Depletion of GSH in glial cells induces neurotoxicity: Relevance to aging and degenerative neurological diseases. FASEB J. 2010, 24, 2533–2545.
- Bahn, G.; Park, J.S.; Yun, U.J.; Lee, Y.J.; Choi, Y.; Park, J.S.; Baek, S.H.; Choi, B.Y.; Cho, Y.S.; Kim, H.K.; et al. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl. Acad. Sci. USA 2019, 116, 12516–12523.
- Deng, Y.; Zhu, J.; Mi, C.; Xu, B.; Jiao, C.; Li, Y.; Xu, D.; Liu, W.; Xu, Z. Melatonin antagonizes Mn-induced oxidative injury through the activation of keap1-Nrf2-ARE signaling pathway in the striatum of mice. Neurotox Res. 2015, 27, 156–171.
- Pontes, G.N.; Cardoso, E.C.; Carneiro-Sampaio, M.M.; Markus, R.P. Pineal melatonin and the innate immune response: The TNF-alpha increase after cesarean section suppresses nocturnal melatonin production. J. Pineal Res. 2007, 43, 365–371.
- Rizor, A.; Pajarillo, E.; Son, D.S.; Aschner, M.; Lee, E. Manganese phosphorylates Yin Yang 1 at serine residues to repress EAAT2 in human H4 astrocytes. Toxicol. Lett. 2022, 355, 41–46.
- Barbosa, A.; Zazula, M.F.; Oliveira, M.C.; Teleken, J.L.; Costa, R.M.; Bonfleur, M.L.; Torrejais, M.M. Maternal exposure to glyphosate-based herbicide promotes changes in the muscle structure of C57BL/6 mice offspring. Anat. Rec. 2022, 305, 3307–3316.
- Pu, Y.; Yang, J.; Chang, L.; Qu, Y.; Wang, S.; Zhang, K.; Xiong, Z.; Zhang, J.; Tan, Y.; Wang, X.; et al. Maternal glyphosate exposure causes autism-like behaviors in offspring through increased expression of soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2020, 117, 11753–11759, Erratum in: Proc. Natl. Acad. Sci. USA 2021, 118, e2100100118.
- Kiernan, M.C.; Ziemann, U.; Eisen, A. Amyotrophic lateral sclerosis: Origins traced to impaired balance between neural excitation and inhibition in the neonatal period. Muscle Nerve 2019, 60, 232–235.
- Ngatuni, D.; Wairagu, P.; Jillani, N.; Isaac, A.O.; Nyariki, J.N. A glyphosate-based herbicide disrupted hematopoiesis and induced organ toxicities, ameliorated by vitamin B12 in a mouse model. Saudi J. Biol. Sci. 2022, 29, 103278.
- Zhang, F.; Zhang, H.B.; Pan, L.P.; Liu, X.; Dou, J.R.; Zhu, B.L. Study on the effect of occupational exposure to glyphosate on blood routine. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2019, 37, 126–129.
- Cocchi, M.; Mondo, E.; Romeo, M.; Traina, G. The Inflammatory Conspiracy in Multiple Sclerosis: A Crossroads of Clues and Insights through Mast Cells, Platelets, Inflammation, Gut Microbiota, Mood Disorders and Stem Cells. Int. J. Mol. Sci. 2022, 23, 3253.
- Fukuda, K.; Kuroda, T.; Tamura, N.; Mita, H.; Miyata, H.; Kasashima, Y. Platelet Lysate Enhances Equine Skeletal Muscle Regeneration in A Bupivacaine-Induced Muscle Injury Model. J. Equine Vet. Sci. 2022, 112, 103892.
- Gholizade, M.; Farhadi, A.; Marzban, M.; Mahmudpour, M.; Nabipour, I.; Kalantarhormozi, M.; Shafiee, G.; Ostovar, A.; Larijani, B.; Darabi, A.H.; et al. Association between platelet, white blood cell count, platelet to white blood cell ratio and sarcopenia in community-dwelling older adults: Focus on Bushehr Elderly Health (BEH) program. BMC Geriatr. 2022, 22, 300.
- Hishizawa, M.; Yamashita, H.; Akizuki, M.; Urushitani, M.; Takahashi, R. TDP-43 levels are higher in platelets from patients with sporadic amyotrophic lateral sclerosis than in healthy controls. Neurochem. Int. 2019, 124, 41–45.
- Sonkar, V.K.; Eustes, A.S.; Ahmed, A.; Jensen, M.; Solanki, M.V.; Swamy, J.; Kumar, R.; Fidler, T.P.; Houtman, J.C.D.; Allen, B.G.; et al. Endogenous SOD2 (Superoxide Dismutase) Regulates Platelet-Dependent Thrombin Generation and Thrombosis During Aging. Arterioscler. Thromb. Vasc. Biol. 2022, 43, 79–91.
- Meek, W.M.; Kucharik, M.P.; Eberlin, C.T.; Naessig, S.A.; Rudisill, S.S.; Martin, S.D. Calf Strain in Athletes. JBJS Rev. 2022, 10, e21.
- Pagan, C.; Goubran-Botros, H.; Delorme, R.; Benabou, M.; Lemière, N.; Murray, K.; Amsellem, F.; Callebert, J.; Chaste, P.; Jamain, S.; et al. Disruption of melatonin synthesis is associated with impaired 14-3-3 and miR-451 levels in patients with autism spectrum disorders. Sci. Rep. 2017, 7, 2096.
- Mesnage, R.; Teixeira, M.; Mandrioli, D.; Falcioni, L.; Ducarmon, Q.R.; Zwittink, R.D.; Mazzacuva, F.; Caldwell, A.; Halket, J.; Amiel, C.; et al. Use of Shotgun Metagenomics and Metabolomics to Evaluate the Impact of Glyphosate or Roundup MON 52276 on the Gut Microbiota and Serum Metabolome of Sprague-Dawley Rats. Environ. Health Perspect. 2021, 129, 17005.
- Puigbò, P.; Leino, L.I.; Rainio, M.J.; Saikkonen, K.; Saloniemi, I.; Helander, M. Does Glyphosate Affect the Human Microbiota? Life 2022, 12, 707.
- Fréville, M.; Estienne, A.; Ramé, C.; Lefort, G.; Chahnamian, M.; Staub, C.; Venturi, E.; Lemarchand, J.; Maximin, E.; Hondelatte, A.; et al. Chronic dietary exposure to a glyphosate-based herbicide results in total or partial reversibility of plasma oxidative stress, cecal microbiota abundance and short-chain fatty acid composition in broiler hens. Front. Physiol. 2022, 13, 974688.
- Ding, W.; Shangguan, Y.; Zhu, Y.; Sultan, Y.; Feng, Y.; Zhang, B.; Liu, Y.; Ma, J.; Li, X. Negative impacts of microcystin-LR and glyphosate on zebrafish intestine: Linked with gut microbiota and microRNAs? Environ. Pollut. 2021, 286, 117685.
- Vazquez-Munoz, R.; Thompson, A.; Russell, J.T.; Sobue, T.; Zhou, Y.; Dongari-Bagtzoglou, A. Insights From the Lactobacillus johnsonii Genome Suggest the Production of Metabolites With Antibiofilm Activity Against the Pathobiont Candida albicans. Front. Microbiol. 2022, 13, 853762.
- Winstone, J.K.; Pathak, K.V.; Winslow, W.; Piras, I.S.; White, J.; Sharma, R.; Huentelman, M.J.; Pirrotte, P.; Velazquez, R. Glyphosate infiltrates the brain and increases pro-inflammatory cytokine TNFα: Implications for neurodegenerative disorders. J. Neuroinflamm. 2022, 19, 193.
- Calingasan, N.Y.; Chen, J.; Kiaei, M.; Beal, M.F. Beta-amyloid 42 accumulation in the lumbar spinal cord motor neurons of amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2005, 19, 340–347.
- Ait-Bali, Y.; Ba-M’hamed, S.; Gambarotta, G.; Sassoè-Pognetto, M.; Giustetto, M.; Bennis, M. Pre- and postnatal exposure to glyphosate-based herbicide causes behavioral and cognitive impairments in adult mice: Evidence of cortical ad hippocampal dysfunction. Arch. Toxicol. 2020, 94, 1703–1723.
- de Castro Vieira Carneiro, C.L.; Chaves, E.M.C.; Neves, K.R.T.; Braga, M.D.M.; Assreuy, A.M.S.; de Moraes, M.E.A.; Aragão, G.F. Behavioral and neuroinflammatory changes caused by glyphosate: Base herbicide in mice offspring. Birth Defects Res. 2022.
- Nozdrenko, D.; Abramchuk, O.; Prylutska, S.; Vygovska, O.; Soroca, V.; Bogutska, K.; Khrapatyi, S.; Prylutskyy, Y.; Scharff, P.; Ritter, U. Analysis of Biomechanical Parameters of Muscle Soleus Contraction and Blood Biochemical Parameters in Rat with Chronic Glyphosate Intoxication and Therapeutic Use of C60 Fullerene. Int. J. Mol. Sci. 2021, 22, 4977.
- Fan, X.; Wang, D.; Shen, X.; Qiu, J.; Wu, L.; Yan, J.; Ji, H. Identification of lncRNA expression profiles and analysis of ceRNA in the hippocampus of perinatal glyphosate-exposed mice. Int. J. Dev. Neurosci. 2021, 81, 312–323.
- Gallegos, C.E.; Bartos, M.; Gumilar, F.; Raisman-Vozari, R.; Minetti, A.; Baier, C.J. Intranasal glyphosate-based herbicide administration alters the redox balance and the cholinergic system in the mouse brain. Neurotoxicology 2020, 77, 205–215.
- Sommansson, A.; Nylander, O.; Sjöblom, M. Melatonin decreases duodenal epithelial paracellular permeability via a nicotinic receptor-dependent pathway in rats in vivo. J. Pineal Res. 2013, 54, 282–291.
- Wang, D.; Wang, X.; Geng, S.; Bi, Z. Axonal regeneration in early stages of sciatic nerve crush injury is enhanced by α7nAChR in rats. Mol. Biol Rep. 2015, 42, 603–609.
- Ojiro, R.; Okano, H.; Takahashi, Y.; Takashima, K.; Tang, Q.; Ozawa, S.; Zou, X.; Woo, G.H.; Shibutani, M. Comparison of the effect of glyphosate and glyphosate-based herbicide on hippocampal neurogenesis after developmental exposure in rats. Toxicology 2022, 483, 153369.
- Masrori, P.; De Schaepdryver, M.; Floeter, M.K.; De Vocht, J.; Lamaire, N.; D’Hondt, A.; Traynor, B.; Poesen, K.; Van Damme, P. Prognostic relationship of neurofilaments, CHIT1, YKL-40 and MCP-1 in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 681–682.
- Baldacci, F.; Lista, S.; Palermo, G.; Giorgi, F.S.; Vergallo, A.; Hampel, H. The neuroinflammatory biomarker YKL-40 for neurodegenerative diseases: Advances in development. Expert Rev. Proteomics 2019, 16, 593–600.
- Kimura, H.; Shimizu, K.; Tanabe, N.; Makita, H.; Taniguchi, N.; Kimura, H.; Suzuki, M.; Abe, Y.; Matsumoto-Sasaki, M.; Oguma, A.; et al. Further evidence for association of YKL-40 with severe asthma airway remodeling. Ann. Allergy Asthma Immunol. 2022, 128, 682–688.e5.
- Tizaoui, K.; Yang, J.W.; Lee, K.H.; Kim, J.H.; Kim, M.; Yoon, S.; Jung, Y.; Park, J.B.; An, K.; Choi, H.; et al. The role of YKL-40 in the pathogenesis of autoimmune diseases: A comprehensive review. Int. J. Biol. Sci. 2022, 18, 3731–3746.
- Tsantilas, P.; Lao, S.; Wu, Z.; Eberhard, A.; Winski, G.; Vaerst, M.; Nanda, V.; Wang, Y.; Kojima, Y.; Ye, J.; et al. Chitinase 3 like 1 is a regulator of smooth muscle cell physiology and atherosclerotic lesion stability. Cardiovasc. Res. 2021, 117, 2767–2780.
- Ferrari-Souza, J.P.; Ferreira, P.C.L.; Bellaver, B.; Tissot, C.; Wang, Y.T.; Leffa, D.T.; Brum, W.S.; Benedet, A.L.; Ashton, N.J.; De Bastiani, M.A.; et al. Astrocyte biomarker signatures of amyloid-β and tau pathologies in Alzheimer’s disease. Mol. Psychiatry 2022, 27, 4781–4789.
- Zhao, T.; Zeng, J.; Xu, Y.; Su, Z.; Chong, Y.; Ling, T.; Xu, H.; Shi, H.; Zhu, M.; Mo, Q.; et al. Chitinase-3 like-protein-1 promotes glioma progression via the NF-κB signaling pathway and tumor microenvironment reprogramming. Theranostics 2022, 12, 6989–7008.
- Hübner, K.; Karwelat, D.; Pietsch, E.; Beinborn, I.; Winterberg, S.; Bedenbender, K.; Benedikter, B.J.; Schmeck, B.; Vollmeister, E. NF-κB-mediated inhibition of microRNA-149-5p regulates Chitinase-3-like 1 expression in human airway epithelial cells. Cell. Signal. 2020, 67, 109498.
- Ma, J.; Zhu, J.; Wang, W.; Ruan, P.; Rajeshkumar, S.; Li, X. Biochemical and Mol. ecular impacts of glyphosate-based herbicide on the gills of common carp. Environ. Pollut. 2019, 252 Pt B, 1288–1300.
- Pandher, U.; Kirychuk, S.; Schneberger, D.; Thompson, B.; Aulakh, G.; Sethi, R.S.; Singh, B. Pulmonary inflammatory response from co-exposure to LPS and glyphosate. Environ. Toxicol. Pharmacol. 2021, 86, 103651.
- Bhat, K.P.; Pelloski, C.E.; Zhang, Y.; Kim, S.H.; deLaCruz, C.; Rehli, M.; Aldape, K.D. Selective repression of YKL-40 by NF-kappaB in glioma cell lines involves recruitment of histone deacetylase-1 and -2. FEBS Lett. 2008, 582, 3193–3200.
- Panther, E.J.; Dodd, W.; Clark, A.; Lucke-Wold, B. Gastrointestinal Microbiome and Neurologic Injury. Biomedicines 2022, 10, 500.

