1. Introduction
It is generally accepted that the pathoetiology and pathophysiology of motor neuron (MNs) loss in amyotrophic lateral sclerosis (ALS) is unknown. As with other neurodegenerative conditions, such as Alzheimer’s disease and Parkinson’s disease, a plethora of biomedical studies have investigated ALS pathophysiology leading to the accumulation of data that are yet to be placed in an accepted pathophysiological framework. Preclinical models have primarily focussed on the use of mutations in Cu/Zn superoxide dismutase (SOD)1, transactive response DNA binding protein 43 kDa (TDP-43) and fused in sarcoma/translated in liposarcoma (FUS), as well as C9ORF72 repeats, which, although infrequent, are the most common ALS susceptibility genes
[1]. Approximately 30 genes, with very low frequency, have also been associated with ALS risk, including Tank-binding kinase 1 (TBK1)
[2] and the CCNF gene (Cyclin-F protein)
[3]. Many of these susceptibility genes are associated with other neurodegenerative disorders, mostly frontotemporal dementia (FTD). Most experimental investigations of susceptibility genes in ALS preclinical models have been carried out in MNs. However, it has been long recognized that the expression of these mutations in rodent astrocytes also has detrimental effects that parallel human ALS pathophysiology, which, in 2007, the authors proposed to indicate astrocyte-specific factors/releases underpin MNs loss in ALS
[4].
Amidst the confusion as to the pathophysiological underpinnings of ALS, a variety of studies have explored, and highlighted, the relevance of different cell types, including microglia
[5], astrocytes
[6], oligodendrocytes
[7], muscles
[8], Schwann cells
[9], the neuromuscular junction (NMJ)
[10], and the gut microbiome
[11]. A recent longitudinal study investigated levels of phosphorylated TDP-43, showing it to be evident in the gut prior to neurological symptom onset by 10 years in one case
[12], indicating a crucial role for alterations in the gut and gut microbiome in ALS pathoetiology, as with many other diverse medical conditions
[13][14][13,14]. Pattle and colleagues found aggregates of pTDP-43 in lamina propria macrophages and dendritic cells as well as myenteric plexus ganglion, neuronal and glial cells
[12]. Aggregates of pTDP-43 were also evident in lymph node parenchyma, endothelial cells, and chondrocytes, indicating wider systemic changes prior to the symptomatology of classical ALS diagnosis.
Recent work indicates a powerful role for heightened CD8
+ t cells and NK cells activation, in association with elevations in interferon (IFN)-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-17A and granzyme B suggestive of heightened cytotoxicity of NK cells and CD8
+ t cells in ALS, which the authors suggest drives MN loss
[15][20]. ALS pathoetiology has classically been thought to be top-down, whilst recent work indicates a strong bottom-up influence, with the latter emphasizing the role of changes in the NMJ and muscle. Recent work has highlighted the interactions of changes in MNs with independent changes in muscle leading to NMJ dysregulation
[8]. However, given the role of the gut in the pathoetiology and the wider systemic changes evident, recent work proposes ALS to be a systemic condition
[16][21].
An array of diverse processes has also been mooted as the primary drivers of ALS symptomatology, including the truncated version of the brain-derived neurotrophic factor (BDNF) receptor, TrkB-TI
[17][22] acid sphingomyelinase
[18][23], ceramide
[19][24], aryl hydrocarbon receptor (AhR)
[20][25], kynurenine
[21][26], tryptophan metabolism
[22][27], melatonin
[23][28], and circadian rhythm disruption
[24][29]. In the FUS model, sleep and circadian disruption precede motor deficits
[25][30], suggesting that circadian dysregulation may be prodromal to classically defined ALS, paralleling similar data in Alzheimer’s disease
[26][31].
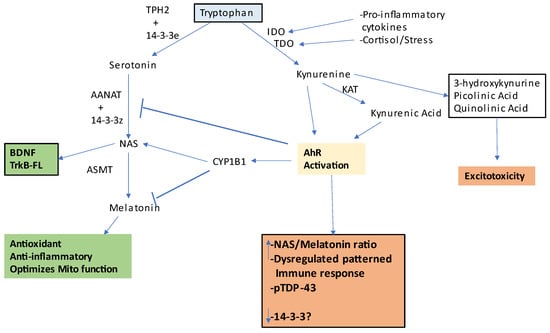
Interestingly, AhR agonists increase soluble and insoluble TDP-43 three-fold
[20][25], suggesting that some of the early TDP-43 increases in the gut and systemically
[12] may be linked to AhR agonism, including from the pro-inflammatory cytokine induction of indoleamine 2,3-dioxygenase (IDO), which converts tryptophan to kynurenine, with kynurenine activating the AhR, in association with a decrease in tryptophan availability for the serotonergic and melatonergic pathway
[27][28][32,33]. See
Figure 1. The AhR has differential impacts on different immune responses, such as the inhibition of NK cell and CD8
+ t cell activation, whilst prolonging macrophage activation. This indicates how alterations in the gut may be intimately linked to the systemic changes in ALS, as in most other medical conditions, and is elaborated below.
Figure 1. This figure shows how tryptophan (light blue shade) can be utilized for beneficial (green) or detrimental (orange) effects in ALS. Tryptophan is converted to serotonin in astrocytes by tryptophan hydroxylase (TPH)2 which has been stabilized by 14-3-3e. Serotonin is converted by AANAT to N-acetylserotonin (NAS) when stabilized by another 14-3-3 isoform and in the presence of acetyl-CoA as a co-substrate. NAS is converted by ASMT to melatonin. NAS can activate the BDNF receptor, TrkB, as well as induce BDNF, whilst melatonin has antioxidant, anti-inflammatory, and mitochondria-optimizing effects. However, in the presence of pro-inflammatory cytokines and stress-associated cortisol-induced IDO and TDO, respectively, tryptophan is converted to kynurenine, which can activate the AhR leading to a number of detrimental effects via kynurenine pathway products that drive excitotoxicity. However, when an active melatonergic pathway is present, AhR-induced CYP1B1 can ‘backward’ convert melatonin to NAS, thereby increasing TrkB-FL activation and BDNF induction. Abbreviations: AANAT: aralkylamine N-acetyltransferase; AhR: aryl hydrocarbon receptor; ASMT: N-acetylserotonin O-methyltransferase; CYP: cytochrome P450; IDO: indoleamine 2,3-dioxygenase; KAT: kynurenine aminotransferase; NAS: N-acetylserotonin; TDO: tryptophan 2,3-dioxygenase; TPH2: tryptophan hydroxylase.
The lack of an integrated conceptualization of ALS pathophysiology means that there is no good treatment target(s). Consequently, ALS treatments are generally very limited in efficacy, typically involving the prescription of Riluzole (to inhibit presynaptic glutamate release)
[29][34], edaravone (primarily as an antioxidant)
[30][35], and selective serotonin reuptake inhibitors (SSRIs) (as MNs first lost are those with most dense serotonin inputs, which show suppressed activity in ALS, with this evident both clinically and preclinically)
[31][36]. Platelet serotonin levels also predict survival duration in ALS patients, highlighting the role of systemic serotonin regulation
[32][37]. However, there is little impact of these medications on ALS life expectancy, with a number of clinical research groups now resorting to finding the best medications to repurpose in the absence of any guiding pathophysiological frame of reference
[33][38]. A wide array of nutriceuticals have been proposed as beneficial in offsetting and slowing ALS-like progression in preclinical models, including green tea and its active polyphenol, epigallocatechin gallate (EGCG)
[34][39], resveratrol, as an AhR antagonist and sirtuin-1 inducer
[35][40], quercetin
[36][41] and sodium butyrate
[37][42], as a histone deacetylase inhibitor (HDACi)
[38][43], as well as the synergistic utility of different combinations of such nutriceuticals
[39][44].
2. Integrating Amyotrophic Lateral SclerosisS Pathophysiology
There is a growing appreciation that many conditions that have been pathophysiologically defined by mutations may be more appropriately seen as disorders in mitochondrial metabolism, driven by mitochondrial ROS-mediated DNA damage, including in cancers
[40][41][146,147], autism spectrum disorders
[42][148], myalgic encephalomyelitis/chronic fatigue syndrome
[43][149], multiple sclerosis
[14] respiratory disorders
[44][150] and many neuropsychiatric conditions
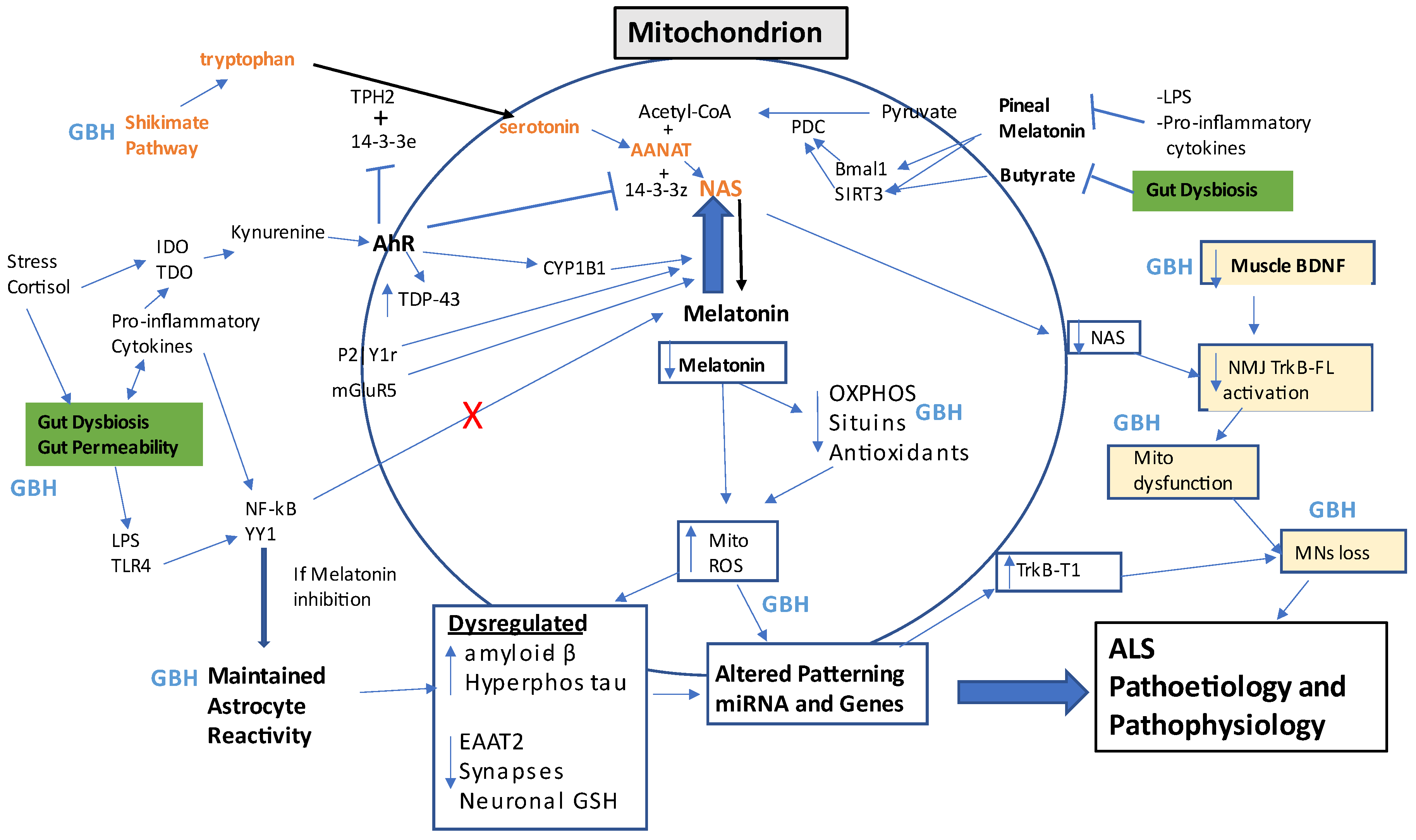
[45][151]. As indicated above, ALS can similarly be defined as arising from alterations in mitochondrial function and the consequences that this has for intercellular processes. The failure to restore intercellular homeostasis arises from dysregulated mitochondrial ROS driving alterations in patterned miRNAs and, consequently, in patterned gene expressions and cellular fluxes. The ability to upregulate the mitochondrial melatonergic pathway is an important aspect of these intracellular and intercellular alterations, providing a novel conceptualization of ALS pathoetiology and pathophysiology. It is proposed that alterations in mitochondrial function occur at three key hubs in ALS, namely the NMJ, glia, and muscle. These key hubs are importantly regulated by systemic processes, namely the gut microbiome/permeability and circadian rhythm, providing a pathophysiological frame of reference upon which it is proposed that GBH can act to increase ALS susceptibility.
This simple outline is complicated by alterations in immune cell function by changes in the gut microbiome/permeability
[13] and circadian rhythm
[46][152], as well as the array of altered fluxes arising from glia, muscle, and MNs/NMJ, mediated by changes in their mitochondria-ROS-patterned miRNAs. This has treatment and preventative implications, as indicated below (Treatment Implications Section}, including the targeting of the mitochondrial melatonergic pathway in key cellular hubs, thereby allowing challenged cells to return closer to their previous homeostatic interactions with other cells. Prevention and treatment derived from such a perspective requires a focus on core aspects of physiological function rather than the plethora of differentially regulated genes and protein effluxes that are currently mooted as treatment targets in ALS and other similarly complex and poorly treated metabolic-systemic medical conditions. Some key aspects are highlighted next (See
Figure 2).
Figure 2. This summary figure shows how many ALS-linked factors can impact mitochondrial function, with relevance across different cell types. Gut dysbiosis is highlighted in green, whilst muscle-NMJ-MNs loss is highlighted in yellow. Gut permeability-associated LPS activates TLR4/NF-kB/YY1 to drive maintained astrocyte activation in the presence of a suppressed melatonergic pathway. The shikimate pathway produces tryptophan (as well as tyrosine and phenylalanine) to drive the serotonin/NAS/melatonin pathway, which requires different 14-3-3 isoforms and acetyl-CoA to be functional. A number of factors can ‘backward’ convert melatonin to NAS, including P2Y1r, mGluR5, and AhR activation. NAS can be beneficial in ALS as it is a BDNF inducer and activator to TrkB-FL. The AhR may also have detrimental effects via 14-3-3 suppression, as well as increasing TDP-43. Gut dysbiosis and pro-inflammatory cytokine suppression of pineal melatonin suppress the capacity of butyrate and melatonin to disinhibit PDC and the PDC conversion of pyruvate to acetyl-CoA, which is a necessary co-substrate of AANAT in the conversion of NAS to melatonin. The suppression of the melatonergic pathway leads to increased oxidants, decreased endogenous antioxidants, OXPHOS, and sirtuins, thereby increasing mitochondrial ROS, ROS-inducing miRNAs, and altered gene patterning, which is a major driver of ALS pathophysiology. The maintained activation of astrocytes decreases neuronal GSH, synapses, and EAAT2 whilst increasing amyloid-β and hyperphosphorylated tau, all of which contribute to ROS-driven changes in patterned miRNAs and gene inductions. The loss of muscle BDNF leads to suboptimal mitochondrial function at the NMJ and MNs loss. All these changes comprise the biological underpinnings of ALS. GBH can have multiple negative impacts, as indicated by points across the figure. Abbreviations: AANAT: aralkylamine N-acetyltransferase; AhR: aryl hydrocarbon receptor; ASMT: N-acetylserotonin O-methyltransferase; BDNF: brain-derived neurotrophic factor; Bmal1: basic helix-loop-helix ARNT Like 1; CYP: cytochrome P450; EAAT: excitatory amino acid transporter; GBH: glyphosate-based herbicides; IDO: indoleamine 2,3-dioxygenase; LPS: lipopolysaccharide; mGluR5: metabotropic glutamate receptor 5; mito: mitochondria; MNs: motor neurons; NAS: N-acetylserotonin; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; NMJ: neuromuscular junction; OXPHOS: oxidative phosphorylation; P2Y1r: purinergic 2Y1 receptor; PDC: pyruvate dehydrogenase complex; ROS: reactive oxygen species; SIRT3: sirtuin-3; TDO: tryptophan 2,3-dioxygenase; TDP-43: transactive response DNA binding protein 43kDa; TLR: toll-like receptor; TPH2: tryptophan hydroxylase; TrkB-FL: tyrosine receptor kinase B-full length; TrkB-T1: tyrosine receptor kinase B-truncated; YYI: yin yang 1.
BDNF regulation in ALS pathophysiology may be intimately linked to alterations in the melatonergic pathway. As noted, a number of miRNAs can regulate the melatonergic pathway, including miR-7, miR-375, miR-451, and miR-709. Some of these miRNAs are also significant regulators of BDNF
[47][48][153,154], suggesting that the important role of BDNF in ALS, especially muscle released BDNF at the NMJ, may be intimately linked to alterations in the mitochondrial melatonergic pathway. Clearly, this will be important to be determined by future research, including the factors that may be differentially regulating these miRNAs across different cell types in ALS patients.
ALS is often diagnosed in late middle age, suggesting an aging-associated factor. As noted, astrocytes are an important hub in ALS pathophysiology. Rodent astrocytes that have been cultured from old (26–29 months) vs. young (4–6 months) mice show a number of significant changes, including slower growth rate, attenuated mitochondrial membrane potential, heightened sensitivity to oxidative challenge, and an enhanced capacity of ATP to induce high Ca2+ responses, which may arise as a consequence of suppressed mitochondrial Ca2+ sequestration
[49][155]. The co-culturing of neurons with aged astrocytes leads to neurons with a heightened sensitivity to oxidative challenge, as to whether these aging-associated changes in astrocytes and their capacity to afford protection to neurons are mediated by a decrease in the astrocyte melatonergic pathway requires investigation. Should the tenfold decrease in pineal melatonin release over human aging (from 18 yrs to 80 yrs) be replicated in astrocytes, this would have significant implications for astrocyte regulation of neuronal function and survival, with clear relevance to how aging associates with neurodegenerative conditions, such as Alzheimer’s disease and Parkinson’s disease as well as ALS.
As noted, the melatonergic pathway is an important determinant of the activation/deactivation of both central and systemic immune cells
[50][51][77,78]. As melatonin increases sirtuins, leading to more optimized mitochondrial function, including the suppression of astrocyte PDK
[52][156], any aging-associated loss of astrocyte melatonin production, release, and autocrine/paracrine effects may be a significant mediator of aging susceptibility to neurodegenerative conditions via impacts on neuronal, including MNs, mitochondrial function. Data showing a 1.5–4.9-fold reduction in hippocampal and saliva sirtuin-1, sirtuin-3, and sirtuin-6 in dementia patients vs. controls
[53][157] may be intimately linked to the suppression of both pineal and local melatonin production. As PDK inhibits PDC, leading to a decreased conversion of pyruvate to acetyl-CoA in astrocytes and a consequent increase in lactate production and release, increased PDK will result in lower levels of astrocyte OXPHOS, coupled with raised levels of lactate production and release
[52][156]. PDK and enhanced glycolysis are crucial for the activation of almost all immune cells due to the heightened metabolic requirements over the course of activation
[54][158], suggesting that aging-associated suppression of the melatonergic pathway in reactive cells, including astrocytes and microglia, may underpin the heightened low-level inflammation often evident in the pathoetiology of neurodegenerative disorders, including ALS.
Melatonin production from the melatonergic pathway would be further suppressed over aging by the enhanced AhR levels and ligands in the course of aging
[55][159], given that the AhR not only backward converts melatonin to NAS but may also act to suppress 14-3-3 and, therefore the stability of the first enzyme in the melatonergic pathway, AANAT
[28][33]. As noted, the suppression of NAS, especially at the NMJ, may be of particular importance in ALS, given the significant role that TrkB-FL activation by BDNF or NAS has on the maintenance of the NMJ, thereby indicating that a decrease in tryptophan availability or uptake, and its conversion by 14-3-3e-stabilized TPH2, may be of particular importance in ALS, especially at the muscle-NMJ interface, with any factors suppressing the availability of NAS likely to increase MNs susceptibility to apoptosis.
Notably, alterations in astrocyte redox status can dramatically alter ionic regulation
[56][160]. These authors showed that astrocyte redox status, primarily GSH regulation, dramatically altered Ca2+ responses to Gq-linked purinergic P2Yr activation, with impacts on IP3-mediated store-operated Ca2+ entry, thereby modulating astrocyte excitability under purinergic stimulation. The inhibition of GSH synthesis in astrocytes and microglia has long been recognized to enhance neuroinflammatory responses
[57][161]. Activation of nuclear factor erythroid-derived 2-related factor 2 (Nrf2), through binding to antioxidant response elements (AREs), inhibits the induction of pro-inflammatory factors, including BACE1 and, therefore, amyloid-β production
[58][162]. This will have consequences for the amyloid-β accumulation around dying MNs. Notably, melatonin activates the Keap1-Nrf2-ARE-GSH pathway in astrocytes
[59][163], indicating that the suppression of pineal melatonin or astrocyte-derived autocrine melatonin, will enhance inflammatory responses to challenge, coupled with alterations in ionic and metabolic regulation. As such, the inhibition of the tryptophan-serotonin-melatonergic pathway by GBH suppression of the shikimate pathway, decreased tryptophan and serotonin availability following gut dysbiosis and increased gut permeability, coupled to 14-3-3 and TPH2 suppression, as well as aging-associated decrements in melatonergic pathway induction, will all contribute to suppressing core aspects of astrocytic regulation of neuronal function and survival. Systemic TNF-α can suppress pineal melatonin production
[60][164], implicating wider systemic inflammation in the melatonin-mediated homeostatic circadian and local regulation of mitochondrial function in immune cells, including glia
[14].
Such processes, as exemplified by astrocytes, are also important at other ALS hubs, including the muscle-NMJ interface and the interactions of the gut microbiome/permeability with intestinal epithelial cells, enteric glial cells, enteric nervous system, and vagal inputs. More specific to astrocytes, the effects of an increased LPS/HMGB1-TLR4/YY1 pathway results in the YY1 suppression of EAAT2 in astrocytes
[61][165], leading to upregulated glutamatergic activity and associated excitotoxicity in MNs, which is the rationale for the use of Riluzole in ALS treatment. This is an important aspect of ALS pathophysiology and may be compensated by an increase in mGluR5 activation, which can ‘backward’ convert melatonin to NAS, thereby having trophic effects at the BDNF receptor, TrkB-FL. However, such compensatory effects of upregulated glutamatergic input onto MNs would be suppressed in the presence of an increased TrkB-T1/TrkB-FL ratio. As such, the rationale for current pharmaceutical treatments in ALS, namely Riluzole and Edaravone, would have their putative therapeutic efficacy (glutamatergic and antioxidant, respectively) intimately linked to the regulation of the mitochondrial melatonergic pathway. As many YY1 transcriptional effects are potentiated by HDAC, the suppression of gut microbiome-derived butyrate and its capacity as an HDACi would be expected to enhance YY1 transcriptions. This again highlights the importance of alterations in the gut microbiome in shaping astrocytic regulation of MNs in the course of ALS pathophysiology.
Glyphosate-Based Herbicides and Integrated Amyotrophic Lateral Sclerosis Pathophysiology
GBH and Integrated ALS Pathophysiology
There is a growing appreciation that GBH may be associated with a wide array of diverse medical conditions, including cancer (especially non-Hodgkin’s lymphoma), neurodegenerative conditions, and neuropsychiatric conditions. As indicated, all of these conditions can be conceptualized as disorders in mitochondrial metabolism
[14][40][41][42][43][44][45][14,146,147,148,149,150,151]. The preclinical data showing GBH exposure prenatally and early postnatally to have impacts on muscles and the NMJ
[62][123], as well as gut microbiome composition and levels of short-chain fatty acids
[63][166], could indicate a role for GBH in the pathoetiology of host of diverse medical conditions, including autism
[63][166] and ALS
[64][167].
As well as data showing GBH to impact an array of different cells and important hubs in ALS pathophysiology, GBH also has significant effects on circulating platelets in preclinical
[65][168] and human investigations
[66][169], which can contribute to alterations in the BBB
[14] and gut microbiome/permeability
[67][170], as well as muscle homeostasis
[68][171] and muscle-atrophy associated sarcopenia
[69][172]. TDP-43 levels are significantly increased in ALS platelets, in correlation with symptom severity
[70][173], whilst the gradual loss of mitochondrial SOD2 in platelets over aging is proposed as a significant factor in an array of aging-associated conditions
[71][174]. Platelet-rich plasma is important in the clinical management of muscle repair, including in professional sports
[72][175]. It will be important for future research to determine whether GBH regulates mitochondrial platelet function and the mitochondrial melatonergic pathway, including via alterations in mitochondrial ROS-induced regulation of 14-3-3-regulating miRNAs, as indicated by data in autism
[73][176]. Some consequences of GBH effects on core hubs in ALS are shown in
Table 1.
Table 1. Summarizes GBH effects in glia, gut, muscles, and NMJ, with associated references. Abbreviations: 5-HT: 5-hydroxytryptamine (serotonin); α7nAChR: alpha 7 nicotinic acetylcholine receptor; BDNF: brain-derived neurotrophic factor; EAAT: excitatory amino acid transporter; GBH: glyphosate-based herbicides; HDACi: histone deacetylase inhibitor; LPS: lipopolysaccharide; MNs: motor neurons; NAS: N-acetylserotonin; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; NMJ: neuromuscular junction; TPH2: tryptophan hydroxylase; TrkB-FL: tyrosine receptor kinase B-full length; TrkB-T1: tyrosine receptor kinase B-truncated.
Data in rodents indicate that early developmental exposure to GBH alters ROS and endogenous antioxidants as well as decreasing mitochondrial SOD2, indicative of enhanced mitochondrial ROS and associated with the patterning of miRNAs and genes induced. Early developmental GBH exposure also has consequences in adulthood in preclinical models, leading to elevations in oxidants (malondialdehyde) and endogenous antioxidant enzymes (catalase) coupled with a decrease in neurogenesis, suggestive of maintained oxidative metabolic challenge
[88][177]. Given the data indicating the detrimental impacts of early developmental exposure to GBH on muscle fiber, nuclei, and fibrosis, as well as on suppressed NMJ levels
[62][123], this is suggestive of early developmental priming that shapes the homeostatic nature achieved, as influenced by alterations in mitochondrial function and the impact this has on inter-cellular communication. As highlighted throughout, the role of the mitochondrial melatonergic pathway and its regulation by environmental factors in development and adulthood, including by GBH, in shaping such altered homeostatic processes will be important to determine.
This frame of reference allows the integration of wider bodies of previously disparate data on ALS, including occupational risk and the growing number of studies highlighting the relevance of variations in mitochondrial function and metabolism across a range of diverse CNS and systemic cells. The emphasis on the role of the mitochondrial melatonergic pathway also allows for the incorporation of recent data highlighting the pathophysiological role of chitinase-3-like protein 1 (CHI3L1), also known as YKL-40, in ALS pathophysiology
[89][178] as well as in an array of other medical conditions, including neurodegenerative disorders more widely
[90][179], asthma
[91][180], autoimmune disease
[92][181] and cardiovascular disorders
[93][182].
YKL-40 is highly expressed in reactivated astrocytes, where it is proposed to mediate the effects of hyperphosphorylated tau in Alzheimer’s disease preclinical models
[94][183]. In glioma, YKL-40 binds NFKB1 to upregulate NF-kB signaling
[95][184], whilst in epithelial cells, NF-kB suppresses miR-149-5p to upregulate YKL-40 following TLR2/3 activation and induction of TNF-α
[96][185]. This requires investigation in glial cells but would suggest that the incapacity of NF-kB to upregulate the melatonergic pathway may allow for interactions of YKL-40 and NF-kB in a positive feedback loop that maintains inflammation when the anti-inflammatory signaling of released melatonin is absent. As GBH increases NF-kB across a range of species and cell types
[97][186] and potentiates LPS in upregulating TLR2
[98][187], GBH may enhance this putative NF-kB/YKL-40 positive feedback loop when the melatonergic pathway is suppressed. This would also have consequences for GBH effects in the gut, and gut-associated regulation of CNS processes, with GBH upregulating circulating LPS and TLR4 activation in astrocytes, leading to NF-kB and YY1 upregulation, with NF-kB effects potentiated by YKL-40 induction in the absence of any intracrine or autocrine melatonin induced by NF-kB. Endogenous TLR4 activators, such as HMGB1 and hsp70, would also be predicted to have significant additive/synergistic interactions with GBH. Such changes would not be expected to be restricted to glia and ALS but evident across neurodegenerative conditions and cancers
[95][184]. This highlights how alterations in the regulation of core processes underpinning mitochondrial function may integrate previously disparate bodies of data on ALS pathophysiology.
How YKL-40 interacts with variations in gut microbiome-derived butyrate in the regulation of NF-kB will be interesting to determine. NF-kB upregulates YKL-40 in glioma cells
[99][188], suggesting that the impacts of variations in the gut microbiome and gut permeability may significantly upregulate NF-kB in CNS cells, which would be potentiated by GBH. In glioblastoma, HDAC1 suppresses NF-kB induction of YKL-40, suggesting that gut microbiome-derived butyrate may potentiate the NF-kB upregulation of YKL-40. Such possible contrasting effects of butyrate will be important to determine in reactive astrocytes and microglia, as any butyrate-mediated upregulation of NF-kB-YKL-40 would suggest gut microbiome-derived butyrate may then contribute to maintained low-level inflammation, as is often thought to underpin most neurodegenerative conditions, in circumstances where the mitochondrial melatonergic pathway is suppressed. As to whether the suppression of glia mitochondria melatonergic pathway parallels infection-driven TNF-α suppression of pineal melatonin production
[60][164] will be interesting to determine, including how this varies over time. Pineal melatonin suppression is proposed to allow local resolution of inflammation, which is then homeostatically reinforced by subsequent pineal melatonin release. Interestingly, circulating pro-inflammatory cytokines also increase gut permeability and associated gut dysbiosis, indicating that CNS and systemic inflammation may co-ordinate pineal melatonin suppression with gut permeability/dysbiosis, allowing the resolution of local inflammation to re-establish the gut barrier, microbiome, and circadian melatonin, which would then reinforce the homeostasis attained following inflammation. Such delayed responses of the gut and pineal gland do not always seem beneficial, as exemplified in preclinical spinal lesion studies
[100][189], where the use of melatonin or butyrate aids recovery, although both are suppressed by the immune cell pro-inflammatory responses to injury. To what extent parallels occur during the course of CNS inflammation as a consequence of microbial type signaling via TLR2/4 will be important to determine.