Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Helder Maiato | -- | 1340 | 2023-01-06 09:40:13 | | | |
| 2 | Catherine Yang | Meta information modification | 1340 | 2023-01-06 10:03:59 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lopes, D.; Maiato, H. The Tubulin Code in Mitosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/39828 (accessed on 07 August 2026).
Lopes D, Maiato H. The Tubulin Code in Mitosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/39828. Accessed August 07, 2026.
Lopes, Danilo, Helder Maiato. "The Tubulin Code in Mitosis" Encyclopedia, https://encyclopedia.pub/entry/39828 (accessed August 07, 2026).
Lopes, D., & Maiato, H. (2023, January 06). The Tubulin Code in Mitosis. In Encyclopedia. https://encyclopedia.pub/entry/39828
Lopes, Danilo and Helder Maiato. "The Tubulin Code in Mitosis." Encyclopedia. Web. 06 January, 2023.
Copy Citation
Mitosis relies on the critical contribution of microtubules, as well as several microtubule-associated proteins (MAPs) and motors, to regulate several key mechanisms underlying the faithful segregation of the genetic material during cell division. The mitotic spindle is an anisotropic and highly heterogeneous structure, with dynamic astral microtubules essentially tyrosinated, in contrast with more stable microtubule subpopulations, such as kinetochore and a fraction of interpolar microtubules, which accumulate detyrosinated, Δ2, acetylated and polyglutamylated tubulin.
microtubule
mitosis
tubulin code
detyrosination
kinetochore
mitotic spindle
centrosome
molecular motors
MAPs
1. A Navigation System Guides Chromosomes to the Spindle Equator
Although tubulin diversity in the mitotic spindle has been recognized for several decades, the respective functional relevance for mitosis remained unclear until recently. One crucial implication of the tubulin code hypothesis is the regulation of MAPs and motors by specific tubulin isotypes and PTMs [1]. Original work in neurons revealed that classic kinesin motors, such as Kinesin-1, are able to recognize and have a preference for microtubules with particular tubulin PTMs, namely detyrosination and acetylation [2][3]. Subsequently, α-tubulin detyrosination was shown to regulate mitotic chromosome congression to the metaphase plate by guiding the microtubule plus-end-directed motor CENP-E/kinesin-7 at kinetochores in human cells [4]. In contrast, the microtubule minus-end-directed motor dynein/dynactin that is also localized at unattached kinetochores [5][6] preferentially associates with tyrosinated microtubules [7][8][9][10], which favor the initiation of motion but are dispensable for subsequent dynein/dynactin processivity [9][10]. Thus, detyrosinated/tyrosinated α-tubulin regulates the activity of opposing kinetochore motors, establishing a navigation system for chromosomes that assists their congression to the spindle equator [11] (Figure 1). Accordingly, during the initial capture of chromosomes, dynein/dynactin counteracts the action of chromokinesins on chromosome arms to move peripheral chromosomes along tyrosinated astral microtubules towards the vicinity of the poles [12]. By transporting peripheral chromosomes to the poles where the microtubule destabilizing activity of Aurora A kinase is higher [13][14], dynein/dynactin prevents the formation of stable end-on kinetochore–microtubule attachments that would otherwise cause the random ejection of polar chromosomes by chromokinesins [11][12]. Once at the poles, Aurora A-mediated phosphorylation activates CENP-E at kinetochores of polar chromosomes [15], thus allowing their transport specifically along detyrosinated spindle microtubules towards the equator. In agreement, recent super-resolution coherent-hybrid stimulated emission depletion microscopy [16] of CENP-E-GFP revealed its exclusive association with stable kinetochore and interpolar microtubule bundles but not with tyrosinated astral microtubules [17]. Curiously, α-tubulin acetylation on K40, which is also enriched on stable spindle microtubules [18], does not interfere with polar chromosome congression [4]. While the potential contribution of other tubulin PTMs to chromosome congression remains unknown, these findings support a robust working model in which tyrosinated/detyrosinated microtubules guide peripheral chromosomes towards the spindle equator.
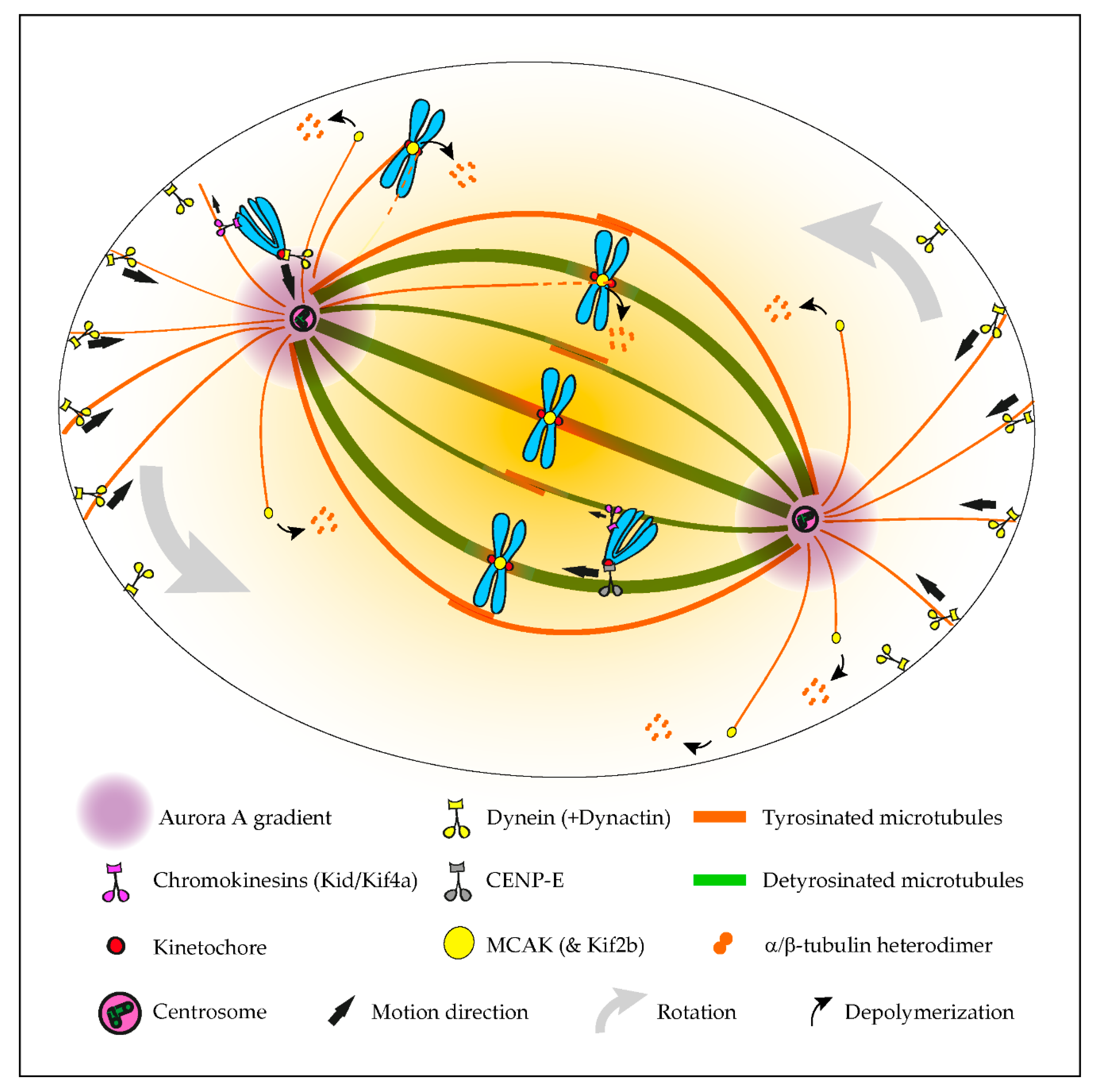
Figure 1. Summary of the established roles of the tubulin code in mitosis. The initial capture of peripheral chromosomes is mediated by dynein/dynactin at kinetochores, upon which, the chromosome is brought to the vicinity of the centrosome by lateral transport along tyrosinated astral microtubules. This prevents the random ejection of the chromosome by the action of Chromokinesins on chromosome arms. Once at the pole, high Aurora A activity prevents the stabilization of end-on kinetochore-microtubule attachments, which otherwise would favor the action of Chromokinesins on chromosome arms. In parallel, Aurora A-mediated phosphorylation activates CENP-E at kinetochores. This initiates transport towards the spindle equator (congression) along stable detyrosinated microtubules. Mitotic centromere-associated kinesin (MCAK) and Kif2b (not depicted) at centromeres and kinetochores are also inhibited by tubulin detyrosination on kinetochore microtubules, allowing the correction of syntelic and merotelic attachments, while preserving correct amphitelic attachments on bi-oriented chromosomes. MCAK at the microtubule plus ends also regulates astral microtubule length to allow interaction with dynein/dynactin at the cortex or cytoplasmic organelles (not depicted), which exerts pulling forces necessary for spindle orientation and positioning.
2. A Mitotic Error Code
The regulation of kinetochore microtubule dynamics is essential for error correction and the maintenance of genome stability, since it allows the establishment of amphitelic kinetochore-MT attachments that lead to chromosome biorientation relative to the spindle poles. Kinesin-13s, such as Kinesin superfamily 2b (Kif2b) and mitotic centromere-associated kinesin (MCAK), promote kinetochore microtubule dynamics, thus playing a key role in the correction of mal-oriented chromosomes with erroneous kinetochore-microtubule attachments (e.g., syntelic, in which both sister kinetochores are oriented towards a single spindle pole, and merotelic, where a single kinetochore is attached with microtubules oriented to both poles) and, ultimately, in the prevention of chromosome mis-segregation [19][20] (Figure 1). In agreement, stimulation of kinetochore microtubule dynamics in otherwise chromosomally unstable cancer cells by increasing kinesin-13 depolymerase activity reestablished chromosomal stability [19][21]. Building on the previous finding that MCAK’s microtubule depolymerizing activity is reduced four-fold in the presence of detyrosinated microtubules in vitro [22][23], it was recently shown that the mitotic error correction activity of MCAK and Kif2b is regulated by α-tubulin detyrosination [24]. Accordingly, the experimental depletion of TTL or overexpression of VASH1-SVBP, which caused a constitutive increase of α-tubulin detyrosination in the vicinity of the kinetochores, compromised error correction, leading to chromosome segregation errors. Importantly, α-tubulin detyrosination specifically impaired the MCAK-based error correction machinery located on centromeres/kinetochores, and it did so without affecting global kinetochore microtubule dynamics, suggesting that mitotic error correction is exquisitely sensitive to the detyrosinated state of α-tubulin that likely occurs at the individual microtubule level. These data support the existence of a “mitotic error code” in which α-tubulin detyrosination/tyrosination signals and regulates MCAK activity at centromeres/kinetochores to discriminate between correct and incorrect kinetochore-MT attachments during mitosis (Figure 1).
Complete centrosome separation before nuclear envelope breakdown prevents subsequent segregation errors and ensures mitotic fidelity [25]. This relies on several elements, including the microtubule motors kinesin-5, required for centrosome separation, and dynein/dynactin, which promote both centrosome separation and positioning [26][27]. Similar to dynein/dynactin, kinesin-5 appears to have increased affinity to tyrosinated dendritic microtubules in neurons [28], but direct evidence from in vitro reconstitution assays is still lacking. Nonetheless, recent work in which centrosome positioning in human mitotic cells was tracked in 3D indicated that centrosome separation at nuclear envelope breakdown is insensitive to the tyrosinated state of α-tubulin [24]. This reinforces the idea that the observed increase in mitotic errors associated with excessive α-tubulin detyrosination is due to the incapacity to correct, rather than an increased propensity to make errors.
3. Role in Mitotic Spindle Orientation and Positioning
Mitotic spindle orientation and positioning in the cell center is essential for accurate cell division and relies on the action of pulling forces on astral microtubules [29]. In particular, dynein/dynactin anchored to cortical proteins or cytoplasmic organelles was shown to play a significant role in spindle orientation/positioning [30][31][32], possibly through its increased affinity to tyrosinated astral microtubules. Indeed, modulation of the α-tubulin tyrosination state, either through TTL knockout [7] or CRISPR/Cas9-mediated editing of the C-terminal tyrosine [32], caused spindle orientation defects. In contrast, an experimental decrease of α-tubulin detyrosination after VASH1/2 silencing increased the depolymerase activity of MCAK, resulting in disoriented spindles, with shorter astral microtubules [33]. Taken together, these observations indicate that the mechanisms behind spindle orientation/positioning rely on the intrinsic nature (i.e., nonmodified) of tyrosinated α-tubulin to allow astral microtubules to establish a correct cell division plane (Figure 1).
4. Roles in Centrosome Structure and Cytokinesis
Tubulin polyglutamylation is highly enriched on centriole microtubules [34][35] and has been proposed to contribute to normal mitosis by maintaining centrosome structure [35][36]. Indeed, recent super-resolution imaging of the centriole structure revealed the specific distribution of polyglutamylation on centriole MTs and suggested a key role for this PTM in ultrastructural organization of specific centriolar proteins [37]. Furthermore, tubulin polyglutamylation promotes the activity of the microtubule-severing enzymes spastin and katanin [38][39][40][41], which are also implicated in cell division. Indeed, their activities regulate several cellular processes that likely impact chromosome segregation fidelity, such as microtubule poleward flux, spindle orientation and length [42][43][44]. Spastin and katanin are also required for the abscission step and completion of cytokinesis [45][46][47]. Like spastin [45] and katanin [47], polyglutamylated tubulin is enriched at the midbody [38], and a tubulin mutation that compromises polyglutamylation (and, possibly, also polyglycylation) in cilia was shown to cause cytokinesis defects [48]. These results suggest that the completion of cytokinesis relies on the regulation of spastin and katanin activities by tubulin polyglutamylation.
References
- Verhey, K.J.; Gaertig, J. The tubulin code. Cell Cycle 2007, 6, 2152–2160.
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172.
- Konishi, Y.; Setou, M. Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat. Neurosci. 2009, 12, 559–567.
- Barisic, M.; Silva e Sousa, R.; Tripathy, S.K.; Magiera, M.M.; Zaytsev, A.V.; Pereira, A.L.; Janke, C.; Grishchuk, E.L.; Maiato, H. Mitosis. Microtubule detyrosination guides chromosomes during mitosis. Science 2015, 348, 799–803.
- Pfarr, C.M.; Coue, M.; Grissom, P.M.; Hays, T.S.; Porter, M.E.; McIntosh, J.R. Cytoplasmic dynein is localized to kinetochores during mitosis. Nature 1990, 345, 263–265.
- Steuer, E.R.; Wordeman, L.; Schroer, T.A.; Sheetz, M.P. Localization of cytoplasmic dynein to mitotic spindles and kinetochores. Nature 1990, 345, 266–268.
- Peris, L.; Thery, M.; Faure, J.; Saoudi, Y.; Lafanechere, L.; Chilton, J.K.; Gordon-Weeks, P.; Galjart, N.; Bornens, M.; Wordeman, L.; et al. Tubulin tyrosination is a major factor affecting the recruitment of cap-gly proteins at microtubule plus ends. J. Cell Biol. 2006, 174, 839–849.
- McKenney, R.J.; Huynh, W.; Tanenbaum, M.E.; Bhabha, G.; Vale, R.D. Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 2014, 345, 337–341.
- McKenney, R.J.; Huynh, W.; Vale, R.D.; Sirajuddin, M. Tyrosination of alpha-tubulin controls the initiation of processive dynein-dynactin motility. EMBO J. 2016, 35, 1175–1185.
- Nirschl, J.J.; Magiera, M.M.; Lazarus, J.E.; Janke, C.; Holzbaur, E.L. Alpha-tubulin tyrosination and clip-170 phosphorylation regulate the initiation of dynein-driven transport in neurons. Cell Rep. 2016, 14, 2637–2652.
- Barisic, M.; Maiato, H. The tubulin code: A navigation system for chromosomes during mitosis. Trends Cell Biol. 2016, 26, 766–775.
- Barisic, M.; Aguiar, P.; Geley, S.; Maiato, H. Kinetochore motors drive congression of peripheral polar chromosomes by overcoming random arm-ejection forces. Nat. Cell Biol. 2014, 16, 1249–1256.
- Chmatal, L.; Yang, K.; Schultz, R.M.; Lampson, M.A. Spatial regulation of kinetochore microtubule attachments by destabilization at spindle poles in meiosis i. Curr. Biol. 2015, 25, 1835–1841.
- Ye, A.A.; Deretic, J.; Hole, C.M.; Hinman, A.W.; Cimini, D.; Welburn, J.P.; Maresca, T.J. Aurora a kinase contributes to a pole-based error correction pathway. Curr. Biol. 2015, 25, 1842–1851.
- Kim, Y.; Holland, A.J.; Lan, W.; Cleveland, D.W. Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of cenp-e. Cell 2010, 142, 444–455.
- Pereira, A.; Sousa, M.; Almeida, A.C.; Ferreira, L.T.; Costa, A.R.; Novais-Cruz, M.; Ferras, C.; Sousa, M.M.; Sampaio, P.; Belsley, M.; et al. Coherent-hybrid sted: High contrast sub-diffraction imaging using a bi-vortex depletion beam. Opt. Express 2019, 27, 8092–8111.
- Steblyanko, Y.; Rajendraprasad, G.; Osswald, M.; Eibes, S.; Jacome, A.; Geley, S.; Pereira, A.J.; Maiato, H.; Barisic, M. Microtubule poleward flux in human cells is driven by the coordinated action of four kinesins. EMBO J. 2020, e105432.
- Wilson, P.J.; Forer, A. Acetylated alpha-tubulin in spermatogenic cells of the crane fly nephrotoma-suturalis-kinetochore microtubules are selectively acetylated. Cell Motil. Cytoskelet. 1989, 14, 237–250.
- Bakhoum, S.F.; Thompson, S.L.; Manning, A.L.; Compton, D.A. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat. Cell Biol. 2009, 11, 27–35.
- Bakhoum, S.F.; Genovese, G.; Compton, D.A. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr. Biol. 2009, 19, 1937–1942.
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472.
- Peris, L.; Wagenbach, M.; Lafanechere, L.; Brocard, J.; Moore, A.T.; Kozielski, F.; Job, D.; Wordeman, L.; Andrieux, A. Motor-dependent microtubule disassembly driven by tubulin tyrosination. J. Cell Biol. 2009, 185, 1159–1166.
- Sirajuddin, M.; Rice, L.M.; Vale, R.D. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol. 2014, 16, 335–344.
- Ferreira, L.T.; Orr, B.; Rajendraprasad, G.; Pereira, A.J.; Lemos, C.; Lima, J.T.; Guasch Boldu, C.; Ferreira, J.G.; Barisic, M.; Maiato, H. Alpha-tubulin detyrosination impairs mitotic error correction by suppressing mcak centromeric activity. J. Cell Biol. 2020, 219, e201910064.
- Silkworth, W.T.; Nardi, I.K.; Paul, R.; Mogilner, A.; Cimini, D. Timing of centrosome separation is important for accurate chromosome segregation. Mol. Biol. Cell 2012, 23, 401–411.
- Nunes, V.; Dantas, M.; Castro, D.; Vitiello, E.; Wang, I.; Carpi, N.; Balland, M.; Piel, M.; Aguiar, P.; Maiato, H.; et al. Centrosome-nuclear axis repositioning drives the assembly of a bipolar spindle scaffold to ensure mitotic fidelity. Mol. Biol. Cell 2020, 31, 1675–1690.
- Raaijmakers, J.A.; van Heesbeen, R.G.H.P.; Meaders, J.L.; Geers, E.F.; Fernandez-Garcia, B.; Medema, R.H.; Tanenbaum, M.E. Nuclear envelope-associated dynein drives prophase centrosome separation and enables eg5-independent bipolar spindle formation. EMBO J. 2012, 31, 4179–4190.
- Kahn, O.I.; Sharma, V.; Gonzalez-Billault, C.; Baas, P.W. Effects of kinesin-5 inhibition on dendritic architecture and microtubule organization. Mol. Biol. Cell 2015, 26, 66–77.
- Siller, K.H.; Doe, C.Q. Spindle orientation during asymmetric cell division. Nat. Cell Biol. 2009, 11, 365–374.
- Kotak, S.; Busso, C.; Gonczy, P. Cortical dynein is critical for proper spindle positioning in human cells. J. Cell Biol. 2012, 199, 97–110.
- Nguyen-Ngoc, T.; Afshar, K.; Gonczy, P. Coupling of cortical dynein and g alpha proteins mediates spindle positioning in caenorhabditis elegans. Nat. Cell Biol. 2007, 9, 1294–1302.
- Barbosa, D.J.; Duro, J.; Prevo, B.; Cheerambathur, D.K.; Carvalho, A.X.; Gassmann, R. Dynactin binding to tyrosinated microtubules promotes centrosome centration in c. Elegans by enhancing dynein-mediated organelle transport. PLoS Genet. 2017, 13, e1006941.
- Liao, S.; Rajendraprasad, G.; Wang, N.; Eibes, S.; Gao, J.; Yu, H.; Wu, G.; Tu, X.; Huang, H.; Barisic, M.; et al. Molecular basis of vasohibins-mediated detyrosination and its impact on spindle function and mitosis. Cell Res. 2019, 29, 533–547.
- Bobinnec, Y.; Moudjou, M.; Fouquet, J.P.; Desbruyeres, E.; Edde, B.; Bornens, M. Glutamylation of centriole and cytoplasmic tubulin in proliferating non-neuronal cells. Cell Motil. Cytoskelet. 1998, 39, 223–232.
- Bobinnec, Y.; Khodjakov, A.; Mir, L.M.; Rieder, C.L.; Edde, B.; Bornens, M. Centriole disassembly in vivo and its effect on centrosome structure and function in vertebrate cells. J. Cell Biol. 1998, 143, 1575–1589.
- Abal, M.; Keryer, G.; Bornens, M. Centrioles resist forces applied on centrosomes during g2/m transition. Biol. Cell 2005, 97, 425–434.
- Mahecic, D.; Gambarotto, D.; Douglass, K.M.; Fortun, D.; Banterle, N.; Ibrahim, K.A.; Le Guennec, M.; Gonczy, P.; Hamel, V.; Guichard, P.; et al. Homogeneous multifocal excitation for high-throughput super-resolution imaging. Nat. Methods 2020, 17, 726–733.
- Lacroix, B.; van Dijk, J.; Gold, N.D.; Guizetti, J.; Aldrian-Herrada, G.; Rogowski, K.; Gerlich, D.W.; Janke, C. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J. Cell Biol. 2010, 189, 945–954.
- Sharma, N.; Bryant, J.; Wloga, D.; Donaldson, R.; Davis, R.C.; Jerka-Dziadosz, M.; Gaertig, J. Katanin regulates dynamics of microtubules and biogenesis of motile cilia. J. Cell Biol. 2007, 178, 1065–1079.
- Shin, S.C.; Im, S.K.; Jang, E.H.; Jin, K.S.; Hur, E.M.; Kim, E.E. Structural and molecular basis for katanin-mediated severing of glutamylated microtubules. Cell Rep. 2019, 26, 1357–1367.
- Valenstein, M.L.; Roll-Mecak, A. Graded control of microtubule severing by tubulin glutamylation. Cell 2016, 164, 911–921.
- Jiang, K.; Rezabkova, L.; Hua, S.S.; Liu, Q.Y.; Capitani, G.; Altelaar, A.F.M.; Heck, A.J.R.; Kammerer, R.A.; Steinmetz, M.O.; Akhmanova, A. Microtubule minus-end regulation at spindle poles by an aspm-katanin complex. Nat. Cell Biol. 2017, 19, 873.
- McNally, K.; Audhya, A.; Oegema, K.; McNally, F.J. Katanin controls mitotic and meiotic spindle length. J. Cell Biol. 2006, 175, 881–891.
- Zhang, D.; Rogers, G.C.; Buster, D.W.; Sharp, D.J. Three microtubule severing enzymes contribute to the "pacman-flux" machinery that moves chromosomes. J. Cell Biol. 2007, 177, 231–242.
- Connell, J.W.; Lindon, C.; Luzio, J.P.; Reid, E. Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic 2009, 10, 42–56.
- Guizetti, J.; Schermelleh, L.; Mantler, J.; Maar, S.; Poser, I.; Leonhardt, H.; Muller-Reichert, T.; Gerlich, D.W. Cortical constriction during abscission involves helices of escrt-iii-dependent filaments. Science 2011, 331, 1616–1620.
- Matsuo, M.; Shimodaira, T.; Kasama, T.; Hata, Y.; Echigo, A.; Okabe, M.; Arai, K.; Makino, Y.; Niwa, S.I.; Saya, H.; et al. Katanin p60 contributes to microtubule instability around the midbody and facilitates cytokinesis in rat cells. PLoS ONE 2013, 8, e80392.
- Thazhath, R.; Liu, C.B.; Gaertig, J. Polyglycylation domain of beta-tubulin maintains axonemal architecture and affects cytokinesis in tetrahymena. Nat. Cell Biol. 2002, 4, 256–259.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Revisions:
2 times
(View History)
Update Date:
06 Jan 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No