 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | David Manuel Pereira De Oliveira | -- | 4650 | 2022-12-30 00:01:19 | | | |
| 2 | Vivi Li | + 268 word(s) | 4918 | 2022-12-30 02:45:53 | | |
Video Upload Options
Enterococcus faecalis, Enterococcus faecium and Staphylococcus aureus are both common commensals and major opportunistic human pathogens. These bacteria have acquired broad resistance to several major classes of antibiotics, including commonly employed glycopeptides. Exemplified by resistance to vancomycin, glycopeptide resistance is mediated through intrinsic gene mutations, and/or transferrable van resistance gene cassette-carrying mobile genetic elements.
1. Introduction
1.1. Enterococcus faecalis and Enterococcus faecium
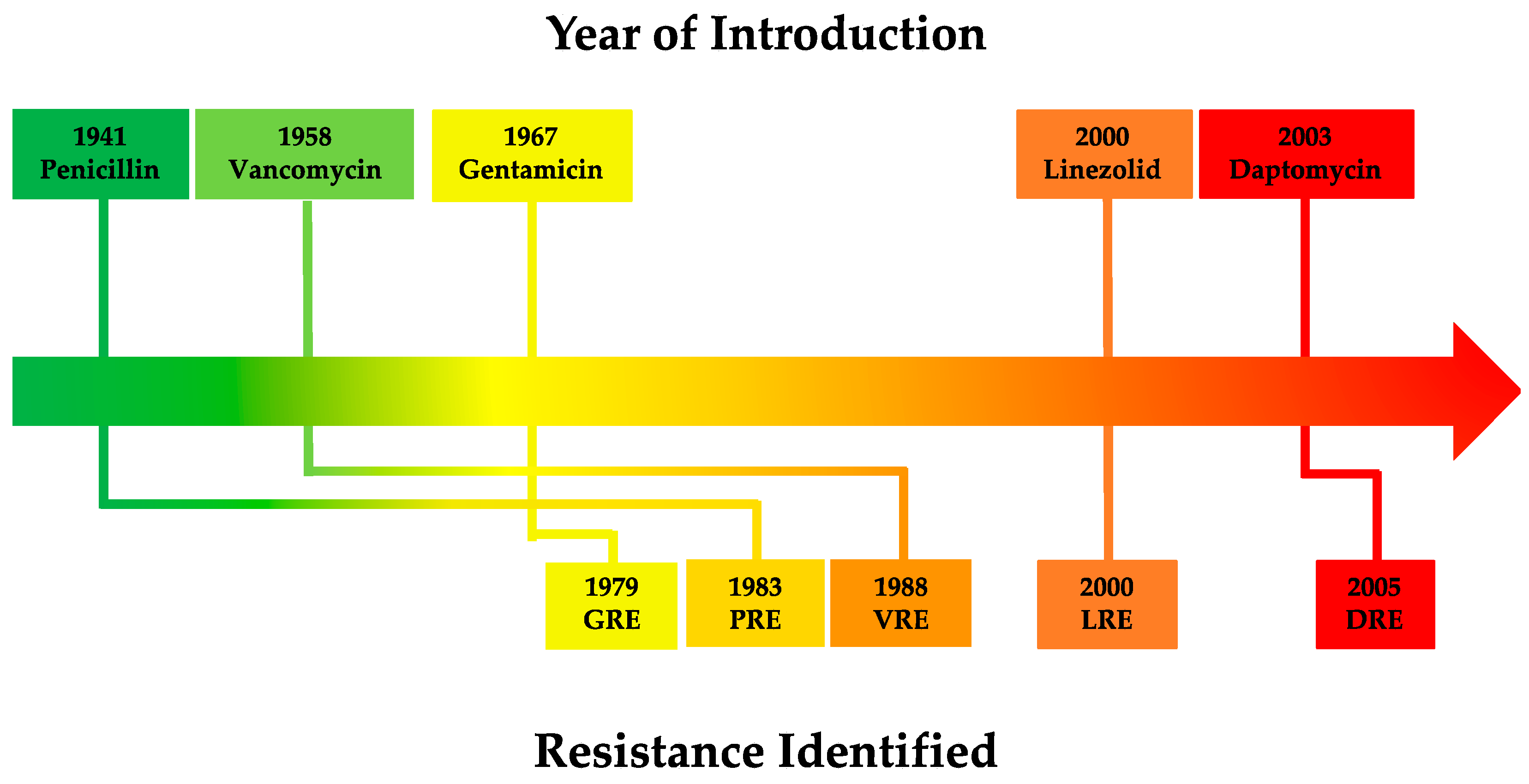
| Antibiotic Class | Resistance Gene(s), Family or Operon | Protein(s) Produced | Mechanism of Action | Gene Location(s) | Enterococcal Mobile Genetic Elements (MGEs) | References |
|---|---|---|---|---|---|---|
| Aminoglycosides | aac | Acetyltransferase | Antibiotic modification and inactivation | Chromosome, plasmid, transposon | Plasmids (P): pIP800, pJH1, pR538-1, pYN134, Inc. 18 Transposons (T): Tn1546, Tn4001, Tn5281, Tn5382, Tn5385 |
[36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55] |
| aad, ant |
Adenylyltransferase | Plasmid, transposon | ||||
| aph | Phosphotransferase | Plasmid, transposon | ||||
| efmM | Methyltransferase | Methylation of 16S rRNA nucleotide; reduction of antibiotic target affinity | Chromosome | - | [56] | |
| Bacitracin, cephalosporins | croRS system | Penicillin-binding protein 5 (PBP5) and others | Cellular signaling in response to cell wall stress; deletion increases cellular susceptibility to antibiotics. Also involved in overexpression of PBP5 | Chromosome | - | [57] |
| β-lactams | blaZ | β-lactamase | Inactivation of β-lactam antibiotics through enzymatic hydrolysis | Chromosome, plasmid, transposon | P: pBEM10 T: Tn5385, Tn552 |
[45][46][58][59][60][61][62] |
| Cephalosporin class β-lactams | pbp5 | Penicillin-binding protein | Reduced antibiotic affinity; enables cell wall cross-linking in the presence of β-lactams | Chromosome, plasmid, transposon | P: The possibility of plasmid-mediated pbp5 transfer has been mentioned. No pbp5-carrying plasmids have been described in E. faecalis or E. faecium, although it has been hypothesized.T: Conjugative transposon CTn5386 | [63][64][65][66][67][68][69] |
| IreK, ireP | IreK—Ser/Thr kinaseireP—protein phosphatase | Part of a signaling transduction pathway that regulates cephalosporin resistance | Chromosome | - | [70] | |
| Chloramphenicol | cat | Chloramphenicol acetyltransferase |
Enzymatic acetylation of chloramphenicol; antibiotic inactivation | Plasmid | P: pRE25, pRUM, pIP501 | [71][72][73][74] |
| Glycopeptides (e.g., vancomycin) | liaFSR, liaXYZ | liaFSR is a regulatory system, with liaXYZ proteins being effector proteins | Modification of the cell membrane and envelope stress response. Modulates cell membrane localization or content, thus altering the antibiotic target site | Chromosome | - | [75][76] |
| cls | Cardiolipin synthase | Involved in cell membrane synthesis; increased cls expression led to membrane modification that impaired antibiotic penetration and activity | Chromosome | - | [77][78][79] | |
| mprF | Bifunctional membrane enzyme involved in phospholipid synthesis and translocation | Increased positive charge of cell membrane and change of membrane fluidity that reduces antibiotic affinity; target modification | Homologs/paralogs described but not extensively studied—gene loci not reported | Unknown | [80][81] | |
| van operon (e.g., vanA) | van operon proteins—refer to Section 2.3 | Reduction of antibiotic affinity through cell wall modification | Chromosome, plasmid, transposon | P: pHKK701, pHKK702, pHK703, pIP816, pIP964, pMG2200, pVEF1 T: Tn1546, Tn1547, Tn1549-like, Tn5482, Tn5506 |
[82][83][84][85][86][87][88][89][90][91] | |
| Lincosamides Oxazolidinones Phenicols Pleuromutilins Streptogramin A |
cfr | rRNA methyltransferase | Methylation of A2503 bacterial 23S rRNA gene; reduced antibiotic affinity to methylated ribosomes | Chromosome, plasmid, transposon |
P: pEF-01 T: IS1216, Tn6218-like |
[92][93][94] |
| optrA a | ABC-F protein | Active dislodgement of antibiotic from its ribosomal target site | Chromosome, plasmid, transposon | P: Inc18, pE349 T: Tn554, Tn6674 |
[95][96][97][98][99] | |
| Linezolid | G2576T | Point mutation in 23S rRNA gene | Ribosomal target modification, reduction of antibiotic affinity | Chromosome | - | [100] |
| Macrolides Lincosamides Streptogramins |
erm | Ribosomal methylase | Methylation of bacterial 23S rRNA domain V; modification of target site and reduced antibiotic binding affinity | Plasmid, transposon | P: pLG2, pRUM-like T: Tn916/Tn1545 |
[45][46][58][101][102][103][104][105][106][107][108] |
| lsa | Efflux pump | Antibiotic efflux | Chromosome, plasmid | P: pMG1-like, pXD4, pY13 | [109][110][111] | |
| msrA b | Chromosome | - | [112][113] | |||
| Phosphonic Acid (e.g., fosfomycin) | fosB | Fosfomycin inactivating enzyme | Mn2+-dependent enzymatic modification and inactivation of fosfomycin | Plasmid, transposon, transferable extrachromosomal intermediate | P: pEMA120 T: ISL3-like, Tn1546-like |
[114][115][116][117] |
| Quinolones | emeA | Efflux pump | Antibiotic efflux | Chromosome | - | [118] |
| gyrA | DNA gyrase mutation | Reduced antibiotic binding affinity | [119][120][121][122] | |||
| parC | Mutation of topoisomerase IV | [119][121][122] | ||||
| qnr | Pentapeptide repeat protein | Protection of DNA gyrase against antibiotic mediated inhibition | [123] | |||
| Streptogramin A | vat | Acetyltransferases | Antibiotic modification and inactivation | Plasmid | P: pAT15, pAT421 | [124][125][126][127][128][129][130] |
| vga | Efflux pump | Antibiotic efflux | Plasmid c | - | [130] | |
| Tetracyclines | tetM, tetO, tetS | Ribosome protection protein | Binding to bacterial ribosome; interference with tetracycline-ribosome binding | Chromosome, plasmid, transposon | P: pDO1–like T: Tn916/Tn1545 family, Tn5397-like |
[45][46][107][108][119][131][132][133][134][135] |
| tetK, tetL | Efflux pump | Antibiotic efflux |
| Antibiotic Class | Resistance gene(s), Family or Operon | Protein(s) produced | Mechanism of Action | Gene Location(s) in S. aureus | Staphylococcal MGEs | References |
|---|---|---|---|---|---|---|
| Aminoglycosides | aac | Acetyltransferase | Antibiotic modification and inactivation | Chromosome, plasmid, transposon | P: pETBTY825, pSK41, pUR1902, pUR2941 T: IS1181, IS1182, Tn4001, Tn5404, Tn5405 Tn554 |
[39][40][41][42][43][44][138][139][140][141][142][143][144][145][146][147][148] |
| aad, ant | Adenylyltransferase | |||||
| aph | Phosphotransferase | |||||
| β-lactams | blaZ | β-lactamase | Inactivation of β-lactam antibiotics through enzymatic hydrolysis | Chromosome, plasmid, transposon | P: pETBTY825, pI258, pI9789 T: Tn552 |
[58][59][145][149][150][151][152][153][154][155][156][157][158][159] |
| Cephalosporins, methicillin |
mecA | Penicillin-binding protein 2a (PBP2a) | Reduced antibiotic affinity; enables cell wall cross-linking in the presence of β-lactams | Chromosome, pathogenicity island (PAI) | PAI: SCCmec | [149][160][161][162][163][164] |
| Chloramphenicol | cat | Chloramphenicol acetyltransferase |
Enzymatic acetylation of chloramphenicol; antibiotic inactivation | Plasmid | P: pC194, pC221, pUB112 | [165][166][167][168][169][170] |
| Glycopeptides (e.g., vancomycin) | cls | Cardiolipin synthase | Involved in cell membrane synthesis; increased cls expression led to membrane modification that impaired antibiotic penetration and activity | Chromosome | - | [171][172][173] |
| mprF | Bifunctional membrane enzyme involved in phospholipid synthesis and translocation | Increased positive charge of cell membrane and change of membrane fluidity that reduces antibiotic affinity; target modification | [174][175][176] | |||
| rpoB | β-subunit of bacterial RNA polymerase | rpoB mutations are frequent in vancomycin-intermediate S. aureus (VISA) strains. They also lead to upregulation of capsule synthesis, attenuated virulence and immune evasion | [177][178][179] | |||
| walKR (also known as yycFG) | Inducible two-component regulator system consisting of a sensor kinase and response regulator | Regulation of cell wall synthesis (thickening), biofilm formation, virulence, immune evasion, autolysis | [180][181][182][183] | |||
| vraS/vraR (vraSR) | Stress sensing and regulatory system that overproduces protective enzymes such as penicillin-binding protein 2 (PBP2) and other cell wall biosynthesis genes in response to antibiotic activity | [177][184][185][186][187][188] | ||||
| van operon (e.g., vanA) | van operon proteins—refer to Section 2.3. | Reduction of antibiotic affinity through cell wall modification | Plasmid, transposon | P: Inc18-like, pLW1043, pSK41-like T: Tn1546 |
[82][189][190][191][192][193] | |
| Fusidic acid | fusA | Mutation to the EF-G ribosome complex | Antibiotic target modification; reduced antibiotic affinity | Chromosome | - | [194] |
| fusB | FusB protein | Prevention of antibiotic interaction with EF-G target site of bacterial ribosome | Chromosome, plasmid, transposon | P: pUB101 T: IS431/257 |
[194][195][196] | |
| fusC | FusC protein | Chromosome, PAI | PAI: SCC476, SCCmecN1, pseudo SCCmec-SCC-SCCCRISPR | [194][197][198][199][200][201] | ||
| Lincosamides Oxazolidinones Phenicols Pleuromutilins Streptogramin A |
cfr | rRNA methyltransferase | Methylation of A2503 bacterial 23S rRNA gene; reduced antibiotic affinity to methylated ribosomes | Chromosome, plasmid, transposon | P: pSCFS3-like, pSCFS7, pSM19035 T: IS21-558, Tn558 |
[202][203][204][205][206][207][208][209][210] |
| optrAa | ABC-F protein | Active dislodgement of antibiotics from the ribosomal target site | Chromosome, transposon | T: Tn6823 | [95][99][211] | |
| Linezolid | G2576T | Point mutation in 23S rRNA gene | Ribosomal target modification, reduction of antibiotic affinity | Chromosome | - | [212] |
| Macrolides Lincosamides Streptogramins (MLS) |
erm | Ribosomal methylase | Methylation of bacterial 23S rRNA domain V; modification of target site and reduced antibiotic binding affinity | Chromosome, plasmid, transposon | P: pE194, pUR1902, pUR2940, pUR2941 T: Tn551, Tn554 |
[148][213][214][215][216][217] |
| lsa | Efflux pump | Antibiotic efflux | Chromosome, plasmid, transposon | P: pV7037 T: Tn560 |
[110][218][219][220] | |
| mdeA | Chromosome | - | [110][221] | |||
| msrA b | Plasmid | P: pETBTY825, pMS97 | [145][222] | |||
| Mupirocin | mupA | Protein modification | Target modification; reduced antibiotic affinity | Chromosome, plasmid, transposon |
P: pJ2947, pXU12 T: IS257 |
[223][224][225][226][227] |
| Phosphonic acid (e.g., Fosfomycin) | fosB | Fosfomycin inactivating enzyme | Mn2+-dependent enzymatic modification and inactivation of fosfomycin | Chromosome, PAI, plasmid, transposon | PAI: SsPI15305 P: pET28, pIP1842 T: IS257-like c |
[117][228][229][230][231][232][233] |
| Quinolones | gyrA, gyrB | DNA gyrase mutation | Reduced antibiotic binding affinity | Chromosome |
- | [213][234] |
| parC, parE | Mutation of topoisomerase IV | [213][234] | ||||
| norA | Efflux pump | Antibiotic efflux | [235] | |||
| qnr | Pentapeptide repeat protein | Protection of DNA gyrase against antibiotic mediated inhibition | Plasmid d | - | [236] | |
| Streptogramin A | vat | Acetyltransferases | Antibiotic modification and inactivation | Chromosomally located conjugative elements, plasmid, transposon | P: pIP524, pIP680, pIP1156, pIP1714 T: Tn5406 |
[126][127][128][129][213][237] |
| vga | Efflux pump | Antibiotic efflux | Chromosome, plasmid, transposon | P: pSA-7, pVGA, pUR2355, pUR4128, pUR3036, pUR3937 T: Tn5406, Tn5406-like, Tn6133 |
[144][237][238][239][240] | |
| Sulfonamides | sulA | Dihydropteroate synthase | Enzymatic overproduction of p-aminobenzoic acid | Chromosome | - | [213] |
| Tetracyclines | tetK, tetL | Efflux pump | Antibiotic efflux | Chromosome, plasmid, transposon | P: pT181, pUR1902, pUR2940, pUR2941, pUSA02 T: Tn1545, Tn5801-like (Tn6014), Tn916 |
[165][193][241][242][243][244][245][246] |
| tetM, tetO, tetS e | Ribosome protection protein | Binding to bacterial ribosome; interference with tetracycline-ribosome binding | ||||
| Trimethoprim | dfrA | Dihydrofolate reductase | Production of trimethroprim-resistant dihydrofolate reductase | Chromosome, plasmid, transposon | P: pSK1, pSK639 T: IS257, Tn4003 |
[245][247][248][249] |
| dfrB | Reduced antibiotic binding affinity | Chromosome | - | [250][251] |
1.2. Staphylococcus aureus
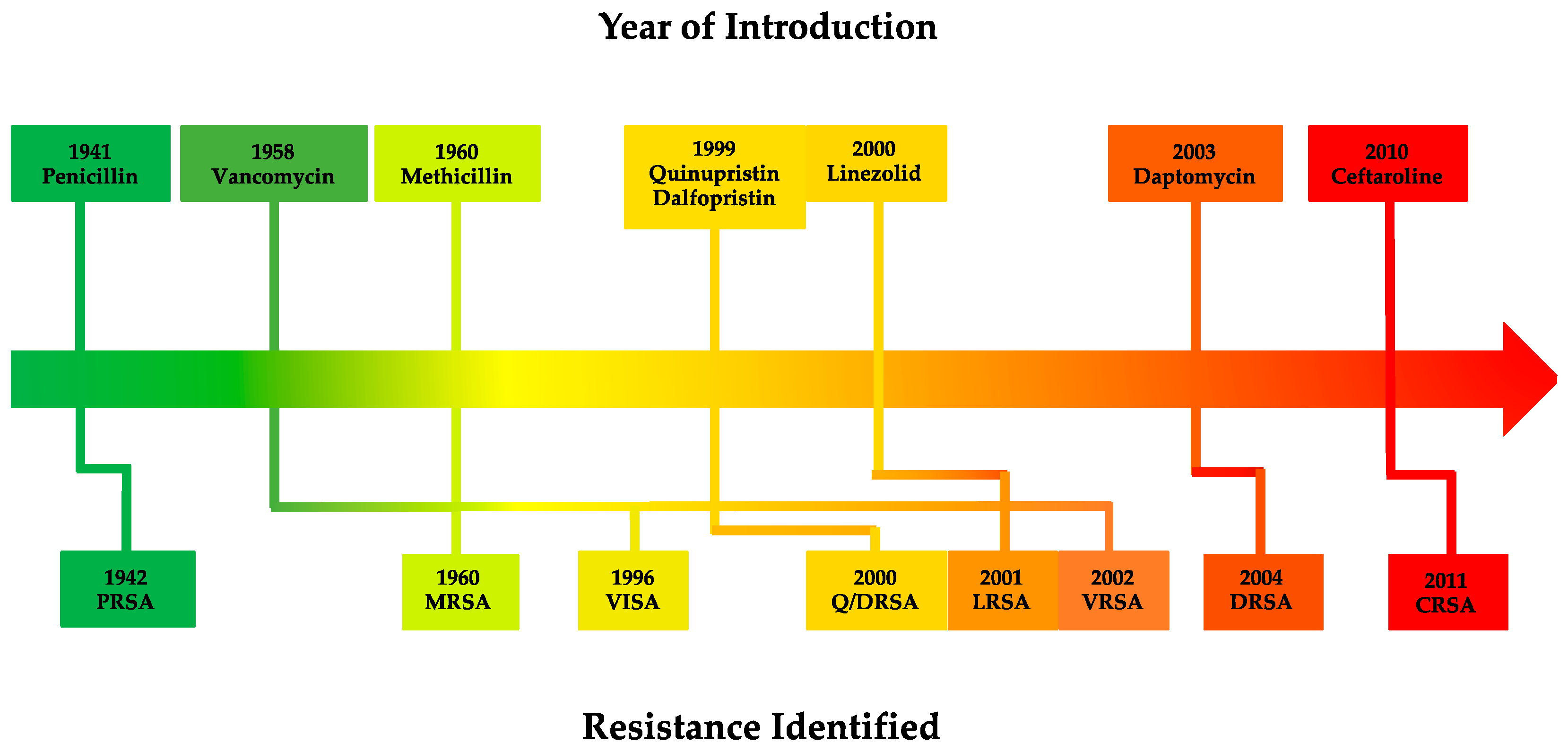
2. Vancomycin
2.1. Discovery and History
2.2. Mechanism of Action
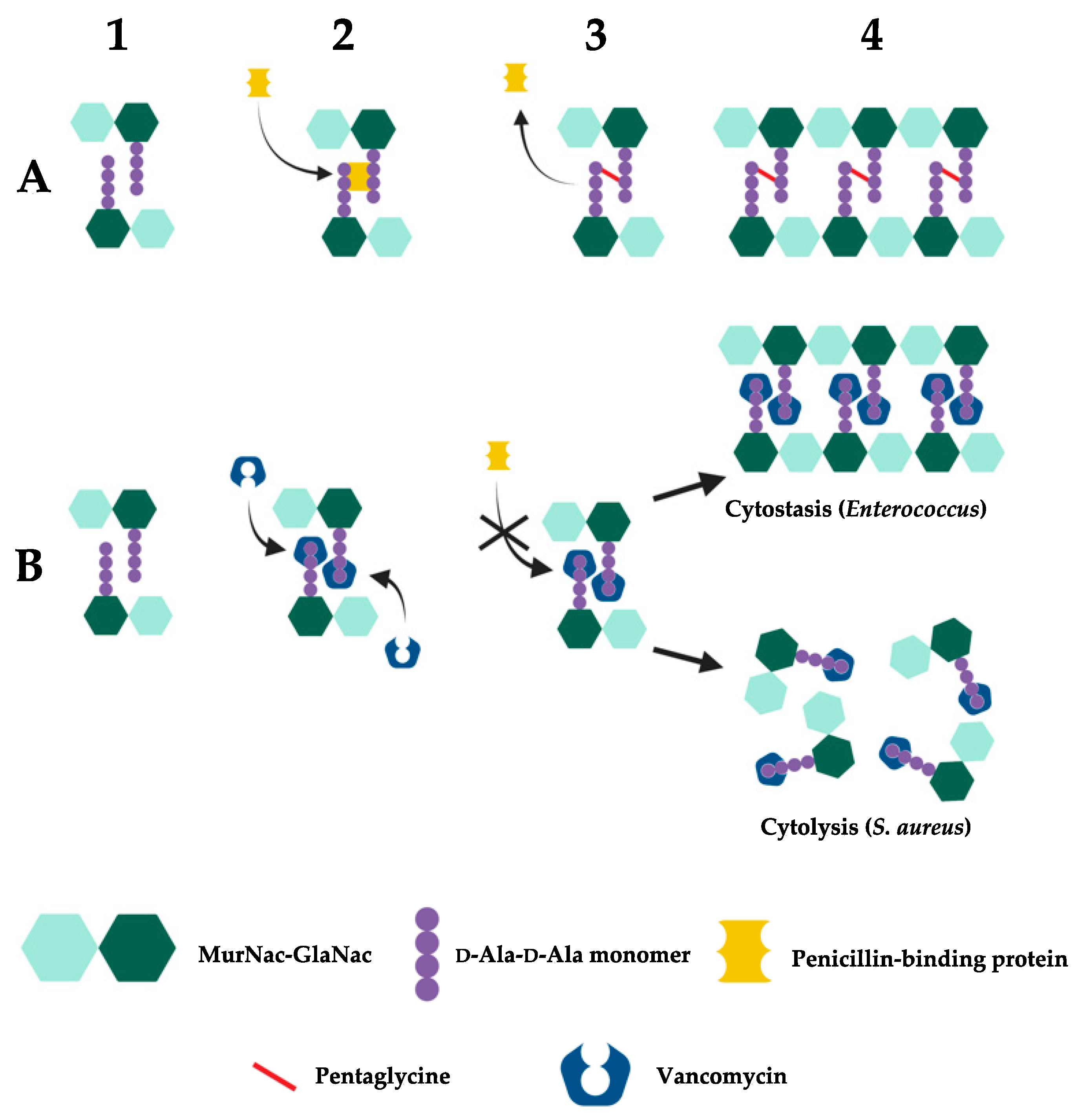
2.3. Vancomycin Resistance in Enterococcus
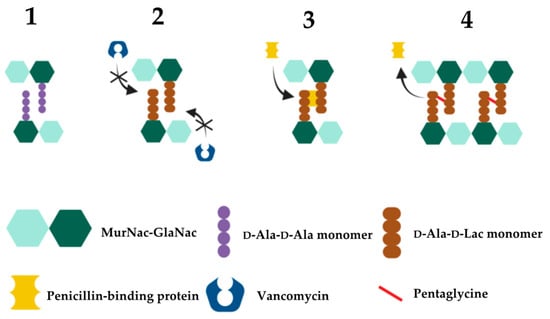
| Gene | Protein/Function | Mechanism of Action | References |
|---|---|---|---|
| D-Ala-D-Lac based resistance (VanA-type resistance)—vanA, vanB, vanD, vanF, vanM gene cassettes High level vancomycin resistance | |||
| vanA1 | Ligase | Catalyses the formation of D-Ala-D-Lac depsipeptides | [192][338] |
| vanH | Dehydrogenase | Catalyses conversion of pyruvate to D-lactate, generating the necessary substrate for D-Ala-D-Lac depsipeptide synthesis | [335][339] |
| vanR/vanS | Regulatory System | The vanR transcription regulator and the vanS sensor kinase comprise the canonical two-component regulatory system that controls vanHAX expression | [340] |
| vanX | Dipeptidase | Cleavage of D-Ala-D-Ala into individual D-Ala residues, thus depleting D-Ala-D-Ala dipeptide substrates from the peptidoglycan synthesis pathway; inhibition of D-Ala-D-Ala synthesis and subsequent loss of binding sites for vancomycin | [341] |
| vanY | Pentapeptidase | D,D-carboxypeptidase activity against D-Ala; reducing availability of D-Ala precursors and therefore favoring the production of peptidoglycan with D-Ala-D-Lac terminals | [342][343][344][345][346] |
| vanZ | Unknown | Currently unknown; vanZ does not appear to be necessary for vancomycin resistance but is required for resistance to the related glycopeptide teicoplanin | [347][348] |
| D-Ala-D-Ser based resistance (VanC-type resistance)—vanC 2, vanE, vanG, vanL, vanN gene cassettes Low level vancomycin resistance | |||
| vanC1 | Ligase | Synthesis of D-Ala-D-Ser peptidoglycan terminals | [336] |
| vanR/vanS | Regulatory system | Two-component regulatory system consisting of the VanR transcription regulator and the VanS sensor kinase | [336] |
| vanT3 | Membrane-bound serine racemase | Catalyses conversion of L-Ser to D-Ser, producing the D-Ser substrates required for D-Ala-D-Ser terminals | [349][350][351][352][353][354][355][356][357][358][359] |
| vanXY3 | Bifunctional dipeptidase/pentapeptidase | Hydrolyses UDP-MurNac-pentapeptides (D-Ala) and D-Ala-D-Ala | [360][361] |
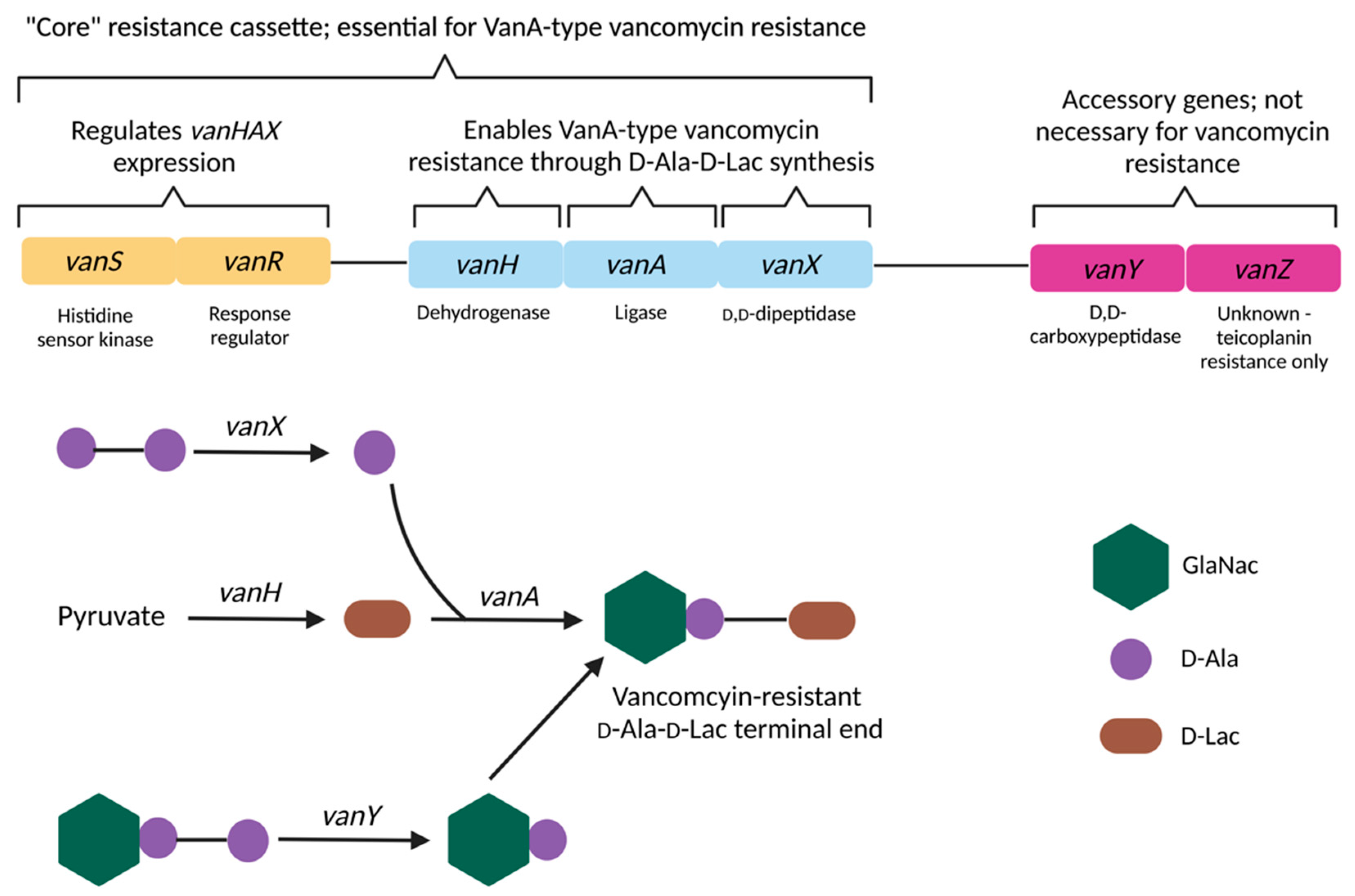
2.4. Vancomycin Resistance in S. aureus
3. Conclusion
E. faecalis, E. faecium and S. aureus are common human commensal organisms with potential to cause serious, life-threatening infections with exceptional intrinsic and acquired antibiotic resistance capabilities that have increased in prevalence globally in recent decades. This is enabled by the emergence and continued dissemination of mobile van resistance cassettes amongst staphylococci and enterococci through MGEs, which confer high-level vancomycin resistance through modification of the D-Ala-D-Ala peptidoglycan terminal ends in addition to endogenous, non-transferable mutations that give rise to intermediate-level resistance in S. aureus. Although other viable antibiotic combinations are still available for vancomycin resistant infections, novel antibiotics, in addition to alternative non-drug antimicrobial strategies will likely be needed to ensure treatment options remain for increasingly drug-resistant infections in the future.
References
- Anderson, A.C.; Jonas, D.; Huber, I.; Karygianni, L.; Wolber, J.; Hellwig, E.; Arweiler, N.; Vach, K.; Wittmer, A.; Al-Ahmad, A. Enterococcus faecalis from Food, Clinical Specimens, and Oral Sites: Prevalence of Virulence Factors in Association with Biofilm Formation. Front. Microbiol. 2016, 6, 1534.
- Jett, B.D.; Huycke, M.M.; Gilmore, M.S. Virulence of enterococci. Clin. Microbiol. Rev. 1994, 7, 462–478.
- Zaheer, R.; Cook, S.R.; Barbieri, R.; Goji, N.; Cameron, A.; Petkau, A.; Polo, R.O.; Tymensen, L.; Stamm, C.; Song, J.; et al. Surveillance of Enterococcus spp. reveals distinct species and antimicrobial resistance diversity across a One-Health continuum. Sci. Rep. 2020, 10, 3937.
- Sghir, A.; Gramet, G.; Suau, A.; Rochet, V.; Pochart, P.; Dore, J. Quantification of Bacterial Groups within Human Fecal Flora by Oligonucleotide Probe Hybridization. Appl. Environ. Microbiol. 2000, 66, 2263–2266.
- Parte, A.C.; Sarda Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Goker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol Microbiol. 2020, 70, 5607–5612.
- Tannock, G.W.; Cook, G. Enterococci as Members of the Intestinal Microflora of Humans. In Enterococci: Pathogenesis, Molecular Biology, and Antibiotic Resistance; Gilmore, M.S., Clewell, D.B., Courvalin, P., Dunny, G.M., Murray, B.E., Rice, L.B., Eds.; ASM Press: Washington, DC, USA, 2002; pp. 101–132.
- Klein, G. Taxonomy, ecology and antibiotic resistance of enterococci from food and the gastro-intestinal tract. Int. J. Food. Microbiol. 2003, 88, 123–131.
- Huycke, M.M.; Sahm, D.F.; Gilmore, M.S. Multiple-drug resistant Enterococci: The nature of the problem and an agenda for the future. Emerg. Infect. Dis. 1998, 4, 239–249.
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharm. Ther. 2015, 40, 277–283.
- Hollenbeck, B.L.; Rice, L.B. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 2012, 3, 421–433.
- Coombs, G.W.; Daley, D.A.; Yee, N.W.T.; Shoby, P.; Mowlaboccus, S. Australian Group on Antimicrobial Resistance (AGAR) Australian Enterococcal Sepsis Outcome Programme (AESOP) Annual Report 2020. Commun. Dis. Intell. 2022, 46.
- Agudelo Higuita, N.I.; Huycke, M.M. Enterococcal Disease, Epidemiology, and Implications for Treatment. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014.
- Kouidhi, B.; Zmantar, T.; Mahdouani, K.; Hentati, H.; Bakhrouf, A. Antibiotic resistance and adhesion properties of oral Enterococci associated to dental caries. BMC Microbiol. 2011, 11, 155.
- Dahlén, G. Role of suspected periodontopathogens in microbiological monitoring of periodontitis. Adv. Dent. Res. 1993, 7, 163–174.
- Rams, T.E.; Feik, D.; Mortensen, J.E.; Degener, J.E.; van Winkelhoff, A.J. Antibiotic Susceptibility of Periodontal Enterococcus faecalis. J. Periodontol. 2013, 84, 1026–1033.
- Pinholt, M.; Østergaard, C.; Arpi, M.; Bruun, N.E.; Schønheyder, H.C.; Gradel, K.O.; Søgaard, M.; Knudsen, J.D. Incidence, clinical characteristics and 30-day mortality of enterococcal bacteraemia in Denmark 2006–2009: A population-based cohort study. Clin. Microbiol. Infect. 2014, 20, 145–151.
- Kao, P.H.N.; Kline, K.A. Jekyll and Mr. Hide: How Enterococcus faecalis Subverts the Host Immune Response to Cause Infection. J. Mol. Biol. 2019, 431, 2932–2945.
- Cattoir, V.; Giard, J.C. Antibiotic resistance in Enterococcus faecium clinical isolates. Expert Rev. Anti-Infect. Ther. 2014, 12, 239–248.
- Hayakawa, K.; Marchaim, D.; Martin, E.T.; Tiwari, N.; Yousuf, A.; Sunkara, B.; Pulluru, H.; Kotra, H.; Hasan, A.; Bheemreddy, S.; et al. Comparison of the clinical characteristics and outcomes associated with vancomycin-resistant Enterococcus faecalis and vancomycin-resistant E. faecium bacteremia. Antimicrob. Agents Chemother. 2012, 56, 2452–2458.
- Garbutt, J.M.; Ventrapragada, M.; Littenberg, B.; Mundy, L.M. Association Between Resistance to Vancomycin and Death in Cases of Enterococcus faecium Bacteremia. Clin. Infect. Dis. 2000, 30, 466–472.
- Giridhara Upadhyaya, P.M.; Ravikumar, K.L.; Umapathy, B.L. Review of virulence factors of Enterococcus: An emerging nosocomial pathogen. Indian J. Med. Microbiol. 2009, 27, 301–305.
- Boneca, I.G.; Chiosis, G. Vancomycin resistance: Occurrence, mechanisms and strategies to combat it. Expert Opin. Ther. Targets 2003, 7, 311–328.
- Dinu, V.; Lu, Y.D.; Weston, N.; Lithgo, R.; Coupe, H.; Channell, G.; Adams, G.G.; Gomez, A.T.; Sabater, C.; Mackie, A.; et al. The antibiotic vancomycin induces complexation and aggregation of gastrointestinal and submaxillary mucins. Sci. Rep. 2020, 10, 960.
- Treitman, A.N.; Yarnold, P.R.; Warren, J.; Noskin, G.A. Emerging incidence of Enterococcus faecium among hospital isolates (1993 to 2002). J. Clin. Microbiol. 2005, 43, 462–463.
- Deshpande, L.M.; Fritsche, T.R.; Moet, G.J.; Biedenbach, D.J.; Jones, R.N. Antimicrobial resistance and molecular epidemiology of vancomycin-resistant enterococci from North America and Europe: A report from the SENTRY antimicrobial surveillance program. Diagn. Microbiol. Infect. Dis. 2007, 58, 163–170.
- Hidron, A.I.; Edwards, J.R.; Patel, J.; Horan, T.C.; Sievert, D.M.; Pollock, D.A.; Fridkin, S.K. NHSN annual update: Antimicrobial-resistant pathogens associated with healthcare-associated infections: Annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol. 2008, 29, 996–1011.
- Zhou, X.; Willems, R.J.L.; Friedrich, A.W.; Rossen, J.W.A.; Bathoorn, E. Enterococcus faecium: From microbiological insights to practical recommendations for infection control and diagnostics. Antimicrob. Resist. Infect. Control 2020, 9, 130.
- Davis, E.; Hicks, L.; Ali, I.; Salzman, E.; Wang, J.; Snitkin, E.; Gibson, K.; Cassone, M.; Mody, L.; Foxman, B. Epidemiology of Vancomycin-Resistant Enterococcus faecium and Enterococcus faecalis Colonization in Nursing Facilities. Open Forum. Infect. Dis. 2020, 7, ofz553.
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2019; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019; p. 150.
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2013; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2013; p. 114.
- Garcia-Solache, M.; Rice, L.B. The Enterococcus: A Model of Adaptability to Its Environment. Clin. Microbiol. Rev. 2019, 32, e00058-18.
- Cheesman, M.J.; Ilanko, A.; Blonk, B.; Cock, I.E. Developing New Antimicrobial Therapies: Are Synergistic Combinations of Plant Extracts/Compounds with Conventional Antibiotics the Solution? Pharmacogn. Rev. 2017, 11, 57–72.
- Levine, D.P. Vancomycin: A History. Clin. Infect. Dis. 2006, 42 (Suppl. S1), S5–S12.
- Moellering, R.C.; Krogstad, D.J.; Greenblatt, D.J. Vancomycin Therapy in Patients with Impaired Renal-Function: A Nomogram for Dosage. Ann. Intern. Med. 1981, 94, 343–346.
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17.
- Chow, J.W. Aminoglycoside Resistance in Enterococci. Clin. Infect. Dis. 2000, 31, 586–589.
- Costa, Y.; Galimand, M.; Leclercq, R.; Duval, J.; Courvalin, P. Characterization of the Chromosomal aac(6’)-Ii Gene-Specific for Enterococcus faecium. Antimicrob. Agents Chemother. 1993, 37, 1896–1903.
- Draker, K.A.; Northrop, D.B.; Wright, G.D. Kinetic Mechanism of the GCN5-Related Chromosomal Aminoglycoside Acetyltransferase AAC(6′)-Ii from Enterococcus faecium: Evidence of Dimer Subunit Cooperativity. Biochemistry 2003, 42, 6565–6574.
- Davies, J.; Wright, G.D. Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol. 1997, 5, 234–240.
- Sundstrom, L.; Radstrom, P.; Swedberg, G.; Skold, O. Site-specific recombination promotes linkage between trimethoprim- and sulfonamide resistance genes. Sequence characterization of dhfrV and sulI and a recombination active locus of Tn21. Mol. Gen. Genet. MGG 1988, 213, 191–201.
- Fling, M.E.; Kopf, J.; Richards, C. Nucleotide sequence of the transposon Tn7 gene encoding an aminoglycoside-modifying enzyme, 3”(9)-O-nucleotidyltransferase. Nucleic Acids Res. 1985, 13, 7095–7106.
- Hollingshead, S.; Vapnek, D. Nucleotide sequence analysis of a gene encoding a streptomycin/spectinomycin adenylyltransferase. Plasmid 1985, 13, 17–30.
- Cameron, F.H.; Obbink, D.J.G.; Ackerman, V.P.; Hall, R.M. Nucleotide sequence of the AAD(2”) aminoglycoside adenylyltransferase determinant aadB. Evolutionary relationship of this region with those surrounding aadA in R538-1 and dhfrll inR388. Nucleic Acids Res. 1986, 14, 8625–8635.
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside Modifying Enzymes. Drug. Resist. Updates 2010, 13, 151–171.
- Rice, L.B.; Carias, L.L. Transfer of Tn5385, a composite, multiresistance chromosomal element from Enterococcus faecalis. J. Bacteriol. 1998, 180, 714–721.
- Rice, L.B. Association of different mobile elements to generate novel integrative elements. Cell. Mol. Life Sci. 2002, 59, 2023–2032.
- Lascols, C.; Legrand, P.; Merens, A.; Leclercq, R.; Muller-Serieys, C.; Drugeon, H.B.; Kitzis, M.D.; Reverdy, M.E.; Roussel-Delvallez, M.; Moubareck, C.; et al. In vitro antibacterial activity of ceftobiprole against clinical isolates from French teaching hospitals: Proposition of zone diameter breakpoints. Int. J. Antimicrob. Agents 2011, 37, 235–239.
- Rice, L.B. Tn916 Family Conjugative Transposons and Dissemination of Antimicrobial Resistance Determinants. Antimicrob. Agents Chemother. 1998, 42, 1871–1877.
- Ono, S.; Muratani, T.; Matsumoto, T. Mechanisms of resistance to imipenem and ampicillin in Enterococcus faecalis. Antimicrob. Agents Chemother. 2005, 49, 2954–2958.
- Chong, Y.P.; Lee, S.O.; Song, E.H.; Lee, E.J.; Jang, E.Y.; Kim, S.H.; Choi, S.H.; Kim, M.N.; Jeong, J.Y.; Woo, J.H.; et al. Quinupristin-dalfopristin versus linezolid for the treatment of vancomycin-resistant Enterococcus faecium bacteraemia: Efficacy and development of resistance. Scand. J. Infect. Dis. 2010, 42, 491–499.
- Berenger, R.; Bourdon, N.; Auzou, M.; Leclercq, R.; Cattoir, V. In vitro activity of new antimicrobial agents against glycopeptide-resistant Enterococcus faecium clinical isolates from France between 2006 and 2008. Med. Mal. Infect. 2011, 41, 405–409.
- Rice, L.B.; Bellais, S.; Carias, L.L.; Hutton-Thomas, R.; Bonomo, R.A.; Caspers, P.; Page, M.G.P.; Gutmann, L. Impact of Specific pbp5 Mutations on Expression of β-Lactam Resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 2004, 48, 3028–3032.
- Arias, C.A.; Singh, K.V.; Panesso, D.; Murray, B.E. Evaluation of ceftobiprole medocaril against Enterococcus faecalis in a mouse peritonitis model. J. Antimicrob. Chemother. 2007, 60, 594–598.
- Daikos, G.L.; Bamias, G.; Kattamis, C.; Zervos, M.J.; Chow, J.W.; Christakis, G.; Petrikkos, G.; Triantafyllopoulou, P.; Alexandrou, H.; Syriopoulou, V. Structures, Locations, and Transfer Frequencies of Genetic Elements Conferring High-Level Gentamicin Resistance in Enterococcus faecalis Isolates in Greece. Antimicrob. Agents Chemother. 2003, 47, 3950–3953.
- Leelaporn, A.; Yodkamol, K.; Waywa, D.; Pattanachaiwit, S. A novel structure of Tn4001-truncated element, type V, in clinical enterococcal isolates and multiplex PCR for detecting aminoglycoside resistance genes. Int. J. Antimicrob. Agents 2008, 31, 250–254.
- Galimand, M.; Schmitt, E.; Panvert, M.; Desmolaize, B.; Douthwaite, S.; Mechulam, Y.; Courvalin, P. Intrinsic resistance to aminoglycosides in Enterococcus faecium is conferred by the 16S rRNA m5C1404-specific methyltransferase EfmM. RNA 2011, 17, 251–262.
- Kellogg, S.L.; Little, J.L.; Hoff, J.S.; Kristich, C.J. Requirement of the CroRS Two-Component System for Resistance to Cell Wall-Targeting Antimicrobials in Enterococcus faecium. Antimicrob. Agents Chemother. 2017, 61, e02461-16.
- Rice, L.B.; Marshall, S.H. Insertions of IS256-like element flanking the chromosomal β-lactamase gene of Enterococcus faecalis CX19. Antimicrob. Agents Chemother. 1994, 38, 693–701.
- Smith, M.C.; Murray, B.E. Sequence analysis of the beta-lactamase repressor from Staphylococcus aureus and hybridization studies with two beta-lactamase-producing isolates of Enterococcus faecalis. Antimicrob. Agents Chemother. 1992, 36, 2265–2269.
- Rice, L.B.; Marshall, S.H. Evidence of Incorporation of the Chromosomal Beta-Lactamase Gene of Enterococcus faecalis CH19 into a Transposon Derived from Staphylococci. Antimicrob. Agents Chemother. 1992, 36, 1843–1846.
- Clewell, D.B.; Weaver, K.E.; Dunny, G.M.; Coque, T.M.; Francia, M.V.; Hayes, F. Extrachromosomal and Mobile Elements in Enterococci: Transmission, Maintenance, and Epidemiology. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014.
- Murray, B.E.; An, F.Y.; Clewell, D.B. Plasmids and Pheromone Response of the β-Lactamase Producer Streptococcus (Enterococcus) faecalis HH22. Antimicrob. Agents Chemother. 1988, 32, 547–551.
- Arbeloa, A.; Segal, H.; Hugonnet, J.E.; Josseaume, N.; Dubost, L.; Brouard, J.P.; Gutmann, L.; Mengin-Lecreulx, D.; Arthur, M. Role of Class A Penicillin-Binding Proteins in PBP5-Mediated β-Lactam Resistance in Enterococcus faecalis. J. Bacteriol. 2004, 186, 1221–1228.
- Carias, L.L.; Rudin, S.D.; Donskey, C.J.; Rice, L.B. Genetic Linkage and Cotransfer of a Novel, vanB-Containing Transposon (Tn5382) and a Low-Affinity Penicillin-Binding Protein 5 Gene in a Clinical Vancomycin-Resistant Enterococcus faecium Isolate. J. Bacteriol. 1998, 180, 4426–4434.
- Rice, L.B.; Carias, L.L.; Rudin, S.; Lakticova, V.; Wood, A.; Hutton-Thomas, R. Enterococcus faecium low-affinity pbp5 is a transferable determinant. Antimicrob. Agents Chemother. 2005, 49, 5007–5012.
- Palmer, K.L.; Godfrey, P.; Griggs, A.; Kos, V.N.; Zucker, J.; Desjardins, C.; Cerqueira, G.; Gevers, D.; Walker, S.; Wortman, J.; et al. Comparative genomics of enterococci: Variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. mBio 2012, 3, e00318-11.
- Garcia-Solache, M.; Lebreton, F.; McLaughlin, R.E.; Whiteaker, J.D.; Gilmore, M.S.; Rice, L.B. Homologous Recombination within Large Chromosomal Regions Facilitates Acquisition of β-Lactam and Vancomycin Resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 2016, 60, 5777–5786.
- Novais, C.; Tedim, A.P.; Lanza, V.F.; Freitas, A.R.; Silveira, E.; Escada, R.; Roberts, A.P.; Al-Haroni, M.; Baguero, F.; Peixe, L.; et al. Co-diversification of Enterococcus faecium Core Genomes and PBP5: Evidences of pbp5 Horizontal Transfer. Front. Microbiol. 2016, 7, 1581.
- Raze, D.; Dardenne, O.; Hallut, S.; Martinez-Bueno, M.; Coyette, J.; Ghuysen, J.M. The gene encoding the low-affinity penicillin-binding protein 3r in Enterococcus hirae S185R is borne on a plasmid carrying other antibiotic resistance determinants. Antimicrob. Agents Chemother. 1998, 42, 534–539.
- Kristich, C.J.; Little, J.L.; Hall, C.L.; Hoff, J.S. Reciprocal Regulation of Cephalosporin Resistance in Enterococcus faecalis. mBio 2011, 2, e00199-11.
- Schwarz, F.V.; Perreten, V.; Teuber, M. Sequence of the 50-kb conjugative multiresistance plasmid pRE25 from Enterococcus faecalis RE25. Plasmid 2001, 46, 170–187.
- Grady, R.; Hayes, F. Axe-Txe, a broad-spectrum proteic toxin-antitoxin system specified by a multidrug-resistant, clinical isolate of Enterococcus faecium. Mol. Microbiol. 2003, 47, 1419–1432.
- Trieu-Cuot, P.; de Cespédès, G.; Bentorcha, F.; Delbos, F.; Gaspar, E.; Horaud, T. Study of heterogeneity of chloramphenicol acetyltransferase (CAT) genes in streptococci and enterococci by polymerase chain reaction: Characterization of a new CAT determinant. Antimicrob. Agents Chemother. 1993, 37, 2593–2598.
- Trieu-Cuot, P.; de Cespedes, G.; Horaud, T. Nucleotide sequence of the chloramphenicol resistance determinant of the streptococcal plasmid pIP501. Plasmid 1992, 28, 272–276.
- Munita, J.M.; Panesso, D.; Diaz, L.; Tran, T.T.; Reyes, J.; Wanger, A.; Murray, B.E.; Arias, C.A. Correlation between Mutations in liaFSR of Enterococcus faecium and MIC of Daptomycin: Revisiting Daptomycin Breakpoints. Antimicrob. Agents Chemother. 2012, 56, 4354–4359.
- Khan, A.; Davlieva, M.; Panesso, D.; Rincon, S.; Miller, W.R.; Diaz, L.; Reyes, J.; Cruz, M.R.; Pemberton, O.; Nguyen, A.H.; et al. Antimicrobial sensing coupled with cell membrane remodeling mediates antibiotic resistance and virulence in Enterococcus faecalis. Proc. Natl. Acad. Sci. USA 2019, 116, 26925–26932.
- Palmer, K.L.; Daniel, A.; Hardy, C.; Silverman, J.; Gilmore, M.S. Genetic Basis for Daptomycin Resistance in Enterococci. Antimicrob. Agents Chemother. 2011, 55, 3345–3356.
- Arias, C.A.; Panesso, D.; McGrath, D.M.; Qin, X.; Mojica, M.F.; Miller, C.; Diaz, L.; Tran, T.T.; Rincon, S.; Barbu, E.M.; et al. Genetic Basis for In Vivo Daptomycin Resistance in Enterococci. N. Engl. J. Med. 2011, 365, 892–900.
- Schlame, M. Thematic Review Series: Glycerolipids. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J. Lipid Res. 2008, 49, 1607–1620.
- Ernst, C.M.; Staubitz, P.; Mishra, N.N.; Yang, S.J.; Hornig, G.; Kalbacher, H.; Bayer, A.S.; Kraus, D.; Peschel, A. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog. 2009, 5, e1000660.
- Bao, Y.; Sakinc, T.; Laverde, D.; Wobser, D.; Benachour, A.; Theilacker, C.; Hartke, A.; Huebner, J. Role of mprF1 and mprF2 in the Pathogenicity of Enterococcus faecalis. PLoS ONE 2012, 7, e38458.
- Cetinkaya, Y.; Falk, P.; Mayhall, C.G. Vancomycin-Resistant Enterococci. Clin. Microbiol. Rev. 2000, 13, 686–707.
- Quintiliani, R.; Courvalin, P. Characterization of Tn1547, a composite transposon flanked by the IS16 and IS256-like elements, that confers vancomycin resistance in Enterococcus faecalis BM4281. Gene 1996, 172, 1–8.
- Handwerger, S.; Skoble, J. Identification of Chromosomal Mobile Element Conferring High-Level Vancomycin Resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 1995, 39, 2446–2453.
- Heaton, M.P.; Discotto, L.F.; Pucci, M.J.; Handwerger, S. Mobilization of vancomycin resistance by transposon-mediated fusion of a VanA plasmid with an Enterococcus faecium sex pheromone-response plasmid. Gene 1996, 171, 9–17.
- Quintiliani, R.; Courvalin, P. Conjugal transfer of the vancomycin resistance determinant vanB between enterococci involves the movement of large genetic elements from chromosome to chromosome. FEMS Microbiol. Lett. 1994, 119, 359–363.
- Arthur, M.; Molinas, C.; Depardieu, F.; Courvalin, P. Characterization of Tn1546, a Tn3-Related Transposon Conferring Glycopeptide Resistance by Synthesis of Depsipeptide Peptidoglycan Precursors in Enterococcus faecium BM4147. J. Bacteriol. 1993, 175, 117–127.
- De Lencastre, H.; Brown, A.E.; Chung, M.; Armstrong, D.; Tomasz, A. Role of Transposon Tn5482 in the Epidemiology of Vancomycin-Resistant Enterococcus faecium in the Pediatric Oncology Unit of a New York City Hospital. Microb. Drug Resist. 1999, 5, 113–129.
- Hung, W.C.; Takano, T.; Higuchi, W.; Iwao, Y.; Khokhlova, O.; Teng, L.J.; Yamamoto, T. Comparative Genomics of Community-Acquired ST59 Methicillin-Resistant Staphylococcus aureus in Taiwan: Novel Mobile Resistance Structures with IS1216V. PLoS ONE 2012, 7, e46987.
- Leclercq, R.; Derlot, E.; Duval, J.; Courvalin, P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N. Engl. J. Med. 1988, 319, 157–161.
- Zheng, B.; Tomita, H.; Inoue, T.; Ike, Y. Isolation of VanB-Type Enterococcus faecalis Strains from Nosocomial Infections: First Report of the Isolation and Identification of the Pheromone-Responsive Plasmids pMG2200, Encoding VanB-Type Vancomycin Resistance and a Bac41-Type Bacteriocin, and pMG2201, Encoding Erythromycin Resistance and Cytolysin (Hly/Bac). Antimicrob. Agents Chemother. 2009, 53, 735–747.
- Shen, J.Z.; Wang, Y.; Schwarz, S. Presence and dissemination of the multiresistance gene cfr in Gram-positive and Gram-negative bacteria. J. Antimicrob. Chemother. 2013, 68, 1697–1706.
- Liu, Y.; Wang, Y.; Wu, C.; Shen, Z.; Schwarz, S.; Du, X.D.; Dai, L.; Zhang, W.; Zhang, Q.; Shen, J. First report of the multidrug resistance gene cfr in Enterococcus faecalis of animal origin. Antimicrob. Agents Chemother. 2012, 56, 1650–1654.
- Kuroda, M.; Sekizuka, T.; Matsui, H.; Suzuki, K.; Seki, H.; Saito, M.; Hanaki, H. Complete Genome Sequence and Characterization of Linezolid-Resistant Enterococcus faecalis Clinical Isolate KUB3006 Carrying a cfr(B)-Transposon on Its Chromosome and optrA-Plasmid. Front. Microbiol. 2018, 9, 2576.
- Ero, R.; Kumar, V.; Su, W.; Gao, Y.G. Ribosome protection by ABC-F proteins—Molecular mechanism and potential drug design. Protein Sci. 2019, 28, 684–693.
- He, T.; Shen, Y.; Schwarz, S.; Cai, J.; Lv, Y.; Li, J.; Feßler, A.T.; Zhang, R.; Wu, C.; Shen, J.; et al. Genetic environment of the transferable oxazolidinone/phenicol resistance gene optrA in Enterococcus faecalis isolates of human and animal origin. J. Antimicrob. Chemother. 2016, 71, 1466–1473.
- Li, D.; Li, X.-Y.; Schwarz, S.; Yang, M.; Zhang, S.-M.; Hao, W.; Du, X.-D. Tn6674 Is a Novel Enterococcal optrA-Carrying Multiresistance Transposon of the Tn554 Family. Antimicrob. Agents Chemother. 2019, 63, e00809-19.
- Almeida, L.M.; Gaca, A.; Bispo, P.M.; Lebreton, F.; Saavedra, J.T.; Silva, R.A.; Basílio-Júnior, I.D.; Zorzi, F.M.; Filsner, P.H.; Moreno, A.M.; et al. Coexistence of the Oxazolidinone Resistance–Associated Genes cfr and optrA in Enterococcus faecalis From a Healthy Piglet in Brazil. Front. Public Health 2020, 8, 518.
- Wang, Y.; Lv, Y.; Cai, J.; Schwarz, S.; Cui, L.; Hu, Z.; Zhang, R.; Li, J.; Zhao, Q.; He, T.; et al. A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. J. Antimicrob. Chemother. 2015, 70, 2182–2190.
- Sinclair, A.; Arnold, C.; Woodford, N. Rapid detection and estimation by pyrosequencing of 23S rRNA genes with a single nucleotide polymorphism conferring linezolid resistance in Enterococci. Antimicrob. Agents Chemother. 2003, 47, 3620–3622.
- Weisblum, B. Erythromycin Resistance by Ribosome Modification. Antimicrob. Agents Chemother. 1995, 39, 577–585.
- Jensen, L.B.; Frimodt-Moller, N.; Aarestrup, F.M. Presence of erm gene classes in Gram-positive bacteria of animal and human origin in Denmark. FEMS Microbiol. Lett. 1999, 170, 151–158.
- Cho, S.H.; Barrett, J.B.; Frye, J.G.; Jackson, C.R. Antimicrobial Resistance Gene Detection and Plasmid Typing Among Multidrug Resistant Enterococci Isolated from Freshwater Environment. Microorganisms 2020, 8, 1338.
- Yao, W.M.; Xu, G.J.; Li, D.Y.; Bai, B.; Wang, H.Y.; Cheng, H.; Zheng, J.X.; Sun, X.; Lin, Z.W.; Deng, Q.W.; et al. Staphylococcus aureus with an erm-mediated constitutive macrolide-lincosamide-streptogramin B resistance phenotype has reduced susceptibility to the new ketolide, solithromycin. BMC Infect. Dis. 2019, 19, 175.
- Bonafede, M.E.; Carias, L.L.; Rice, L.B. Enterococcal transposon Tn5384: Evolution of a composite transposon through cointegration of enterococcal and staphylococcal plasmids. Antimicrob. Agents Chemother. 1997, 41, 1854–1858.
- Laverde Gomez, J.A.; Hendrickx, A.P.A.; Willems, R.J.; Top, J.; Sava, I.; Huebner, J.; Witte, W.; Werner, G. Intra- and Interspecies Genomic Transfer of the Enterococcus faecalis Pathogenicity Island. PLoS ONE 2011, 6, e16720.
- Morroni, G.; Di Cesare, A.; Di Sante, L.; Brenciani, A.; Vignaroli, C.; Pasquaroli, S.; Giovanetti, E.; Sabatino, R.; Rossi, L.; Magnani, M.; et al. Enterococcus faecium ST17 from Coastal Marine Sediment Carrying Transferable Multidrug Resistance Plasmids. Microb. Drug Resist. 2016, 22, 523–530.
- De Leener, E.; Martel, A.; Decostere, A.; Haesebrouck, F. Distribution of the erm(B) Gene, Tetracycline Resistance Genes, and Tn1545-like Transposons in Macrolide- and Lincosamide-Resistant Enterococci from Pigs and Humans. Microb. Drug Resist. 2004, 10, 341–345.
- Yan, X.-M.; Wang, J.; Tao, X.-X.; Jia, H.-B.; Meng, F.-L.; Yang, H.; You, Y.-H.; Zheng, B.; Hu, Y.; Bu, X.-X.; et al. A Conjugative MDR pMG1-Like Plasmid Carrying the lsa(E) Gene of Enterococcus faecium With Potential Transmission to Staphylococcus aureus. Front. Microbiol. 2021, 12, 667415.
- Poole, K. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 2005, 56, 20–51.
- Li, X.-S.; Dong, W.-C.; Wang, X.-M.; Hu, G.-Z.; Wang, Y.-B.; Cai, B.-Y.; Wu, C.-M.; Wang, Y.; Du, X.-D. Presence and genetic environment of pleuromutilin–lincosamide–streptogramin A resistance gene lsa(E) in enterococci of human and swine origin. J. Antimicrob. Chemother. 2013, 69, 1424–1426.
- Zhao, C.; Hartke, A.; La Sorda, M.; Posteraro, B.; Laplace, J.M.; Auffray, Y.; Sanguinetti, M. Role of Methionine Sulfoxide Reductases A and B of Enterococcus faecalis in Oxidative Stress and Virulence. Infect. Immun. 2010, 78, 3889–3897.
- Portillo, A.; Ruiz-Larrea, F.; Zarazaga, M.; Alonso, A.; Martinez, J.L.; Torres, C. Macrolide Resistance Genes in Enterococcus spp. Antimicrob. Agents Chemother. 2000, 44, 967–971.
- Sun, L.Y.; Zhang, P.; Qu, T.T.; Chen, Y.; Hua, X.T.; Shi, K.R.; Yu, Y.S. Identification of Novel Conjugative Plasmids with Multiple Copies of fosB that Confer High-Level Fosfomycin Resistance to Vancomycin-Resistant Enterococci. Front. Microbiol. 2017, 8, 1541.
- Qu, T.T.; Shi, K.R.; Ji, J.S.; Yang, Q.; Du, X.X.; Wei, Z.Q.; Yu, Y.S. Fosfomycin resistance among vancomycin-resistant enterococci owing to transfer of a plasmid harbouring the fosB gene. Int. J. Antimicrob. Agents 2014, 43, 361–365.
- Xu, X.G.; Chen, C.H.; Lin, D.F.; Guo, Q.L.; Hu, F.P.; Zhu, D.M.; Li, G.H.; Wang, M.G. The Fosfomycin Resistance Gene fosB3 Is Located on a Transferable, Extrachromosomal Circular Intermediate in Clinical Enterococcus faecium Isolates. PLoS ONE 2013, 8, e78106.
- Thompson, M.K.; Keithly, M.E.; Goodman, M.C.; Hammer, N.D.; Cook, P.D.; Jagessar, K.L.; Harp, J.; Skaar, E.P.; Armstrong, R.N. Structure and Function of the Genomically Encoded Fosfomycin Resistance Enzyme, FosB, from Staphylococcus aureus. Biochemistry 2014, 53, 755–765.
- Jonas, B.M.; Murray, B.E.; Weinstock, G.M. Characterization of emeA, a norA Homolog and Multidrug Resistance Efflux Pump, in Enterococcus faecalis. Antimicrob. Agents Chemother. 2001, 45, 3574–3579.
- Mbanga, J.; Amoako, D.G.; Abia, A.L.K.; Allam, M.; Ismail, A.; Essack, S.Y. Genomic Analysis of Enterococcus spp. Isolated From a Wastewater Treatment Plant and Its Associated Waters in Umgungundlovu District, South Africa. Front. Microbiol. 2021, 12, 648454.
- Oana, K.; Okimura, Y.; Kawakami, Y.; Hayashida, N.; Shimosaka, M.; Okazaki, M.; Hayashi, T.; Ohnishi, M. Physical and genetic map of Enterococcus faecium ATCC19434 and demonstration of intra- and interspecific genomic diversity in enterococci. FEMS Microbiol. Lett. 2002, 207, 133–139.
- Petersen, A.; Jensen, L.B. Analysis of gyrA and parC mutations in enterococci from environmental samples with reduced susceptibility to ciprofloxacin. FEMS Microbiol. Lett. 2004, 231, 73–76.
- Kanematsu, E.; Deguchi, T.; Yasuda, M.; Kawamura, T.; Nishino, Y.; Kawada, Y. Alterations in the GyrA subunit of DNA gyrase and the ParC subunit of DNA topoisomerase IV associated with quinolone resistance in Enterococcus faecalis. Antimicrob. Agents Chemother. 1998, 42, 433–435.
- Arsène, S.; Leclercq, R. Role of a qnr-Like Gene in the Intrinsic Resistance of Enterococcus faecalis to Fluoroquinolones. Antimicrob. Agents Chemother. 2007, 51, 3254–3258.
- Simjee, S.; White, D.G.; Wagner, D.D.; Meng, J.; Qaiyumi, S.; Zhao, S.; McDermott, P.F. Identification of vat(E) in Enterococcus faecalis Isolates from Retail Poultry and Its Transferability to Enterococcus faecium. Antimicrob. Agents Chemother. 2002, 46, 3823–3828.
- Soltani, M.; Beighton, D.; Philpott-Howard, J.; Woodford, N. Mechanisms of Resistance to Quinupristin-Dalfopristin among Isolates of Enterococcus faecium from Animals, Raw Meat, and Hospital Patients in Western Europe. Antimicrob. Agents Chemother. 2000, 44, 433–436.
- Allignet, J.; Elsolh, N. Diversity among the Gram-Positive Acetyltransferases Inactivating Streptogramin a and Structurally Related-Compounds and Characterization of a New Staphylococcal Determinant, vatB. Antimicrob. Agents Chemother. 1995, 39, 2027–2036.
- Allignet, J.; Liassine, N.; El Solh, N. Characterization of a Staphylococcal Plasmid Related to pUB110 and Carrying Two Novel Genes, vatC and vgbB, Encoding Resistance to Streptogramins A and B and Similar Antibiotics. Antimicrob. Agents Chemother. 1998, 42, 1794–1798.
- Allignet, J.; Loncle, V.; Simenel, C.; Delepierre, M.; Elsolh, N. Sequence of a staphylococcal gene, vat, encoding an acetyltransferase inactivating the A-type compounds of virginiamycin-like antibiotics. Gene 1993, 130, 91–98.
- Roberts, M.C.; Sutcliffe, J.; Courvalin, P.; Jensen, L.B.; Rood, J.; Seppala, H. Nomenclature for Macrolide and Macrolide-Lincosamide-Streptogramin B Resistance Determinants. Antimicrob. Agents Chemother. 1999, 43, 2823–2830.
- Jung, Y.H.; Shin, E.S.; Kim, O.; Yoo, J.S.; Lee, K.M.; Yoo, J.I.; Chung, G.T.; Lee, Y.S. Characterization of two newly identified genes, vgaD and vatH, conferring resistance to streptogramin A in Enterococcus faecium. Antimicrob. Agents Chemother. 2010, 54, 4744–4749.
- Li, W.; Atkinson, G.C.; Thakor, N.S.; Allas, U.; Lu, C.C.; Chan, K.Y.; Tenson, T.; Schulten, K.; Wilson, K.S.; Hauryliuk, V.; et al. Mechanism of tetracycline resistance by ribosomal protection protein Tet(O). Nat. Commun. 2013, 4, 1477.
- Molale, L.G.; Bezuidenhout, C.C. Antibiotic resistance, efflux pump genes and virulence determinants in Enterococcus spp. from surface water systems. Environ. Sci. Pollut. Res. 2016, 23, 21501–21510.
- Agerso, Y.; Pedersen, A.G.; Aarestrup, F.M. Identification of Tn5397-like and Tn916-like transposons and diversity of the tetracycline resistance gene tet(M) in enterococci from humans, pigs and poultry. J. Antimicrob. Chemother. 2006, 57, 832–839.
- You, Y.Q.; Hilpert, M.; Ward, M.J. Detection of a Common and Persistent tet(L)-Carrying Plasmid in Chicken-Waste-Impacted Farm Soil. Appl. Environ. Microbiol. 2012, 78, 3203–3213.
- Huys, G.; D’Haene, K.; Collard, J.M.; Swings, J. Prevalence and Molecular Characterization of Tetracycline Resistance in Enterococcus Isolates from Food. Appl. Environ. Microbiol. 2004, 70, 1555–1562.
- Crowe-McAuliffe, C.; Murina, V.; Turnbull, K.J.; Huch, S.; Kasari, M.; Takada, H.; Nersisyan, L.; Sundsfjord, A.; Hegstad, K.; Atkinson, G.C.; et al. Structural basis for PoxtA-mediated resistance to phenicol and oxazolidinone antibiotics. Nat. Commun. 2022, 13, 1860.
- Lim, J.-A.; Kwon, A.-R.; Kim, S.-K.; Chong, Y.; Lee, K.; Choi, E.-C. Prevalence of resistance to macrolide, lincosamide and streptogramin antibiotics in Gram-positive cocci isolated in a Korean hospital. J. Antimicrob. Chemother. 2002, 49, 489–495.
- Rouch, D.A.; Byrne, M.E.; Kong, Y.C.; Skurray, R.A. The aacA-aphD gentamicin and kanamycin resistance determinant of Tn4001 from Staphylococcus aureus: Expression and nucleotide sequence analysis. J. Gen. Microbiol. 1987, 133, 3039–3052.
- Lyon, B.R.; May, J.W.; Skurray, R.A. Tn4001—A Gentamicin and Kanamycin Resistance Transposon in Staphylococcus aureus. Mol. Gen. Genet. 1984, 193, 554–556.
- Trieu-Cuot, P.; Courvalin, P. Nucleotide sequence of the Streptococcus faecalis plasmid gene encoding the 3’5”-aminoglycoside phosphotransferase type III. Gene 1983, 23, 331–341.
- Ferretti, J.J.; Gilmore, K.S.; Courvalin, P. Nucleotide sequence analysis of the gene specifying the bifunctional 6’-aminoglycoside acetyltransferase 2”-aminoglycoside phosphotransferase enzyme in Streptococcus faecalis and identification and cloning of gene regions specifying the two activities. J. Bacteriol. 1986, 167, 631–638.
- Murphy, E. Nucleotide sequence of a spectinomycin adenyltransferase AAD(9) determinant from Staphylococcus aureus and its relationship to AAD(3”) (9). Mol. Gen. Genet. 1985, 200, 33–39.
- Kayser, F.H.; Homberger, F.; Devaud, M. Aminocyclitol-Modifying Enzymes Specified by Chromosomal Genes in Staphylococcus aureus. Antimicrob. Agents Chemother. 1981, 19, 766–772.
- Schwendener, S.; Perreten, V. New Transposon Tn6133 in Methicillin-Resistant Staphylococcus aureus ST398 Contains vga(E), a Novel Streptogramin A, Pleuromutilin, and Lincosamide Resistance Gene. Antimicrob. Agents Chemother. 2011, 55, 4900–4904.
- Hisatsune, J.; Hirakawa, H.; Yamaguchi, T.; Fudaba, Y.; Oshima, K.; Hattori, M.; Kato, F.; Kayama, S.; Sugai, M. Emergence of Staphylococcus aureus Carrying Multiple Drug Resistance Genes on a Plasmid Encoding Exfoliative Toxin B. Antimicrob. Agents Chemother. 2013, 57, 6131–6140.
- Schmitz, F.-J.; Fluit, A.C.; Gondolf, M.; Beyrau, R.; Lindenlauf, E.; Verhoef, J.; Heinz, H.-P.; Jones, M.E. The prevalence of aminoglycoside resistance and corresponding resistance genes in clinical isolates of staphylococci from 19 European hospitals. J. Antimicrob. Chemother. 1999, 43, 253–259.
- Derbise, A.; Dyke, K.G.; el Solh, N. Characterization of a Staphylococcus aureus transposon, Tn5405, located within Tn5404 and carrying the aminoglycoside resistance genes, aphA-3 and aadE. Plasmid 1996, 35, 174–188.
- Gómez-Sanz, E.; Kadlec, K.; Feßler, A.T.; Zarazaga, M.; Torres, C.; Schwarz, S. Novel erm(T)-carrying multiresistance plasmids from porcine and human isolates of methicillin-resistant Staphylococcus aureus ST398 that also harbor cadmium and copper resistance determinants. Antimicrob. Agents Chemother. 2013, 57, 3275–3282.
- Hackbarth, C.J.; Chambers, H.F. blaI and blaR1 Regulate β-Lactamase and PBP 2a Production in Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 1993, 37, 1144–1149.
- Pence, M.A.; Haste, N.M.; Meharena, H.S.; Olson, J.; Gallo, R.L.; Nizet, V.; Kristian, S.A. Beta-Lactamase Repressor BlaI Modulates Staphylococcus aureus Cathelicidin Antimicrobial Peptide Resistance and Virulence. PLoS ONE 2015, 10, e0136605.
- Zscheck, K.K.; Murray, B.E. Genes Involved in the Regulation of β-Lactamase Productionin Enterococci and Staphylococci. Antimicrob. Agents Chemother. 1993, 37, 1966–1970.
- Lyon, B.R.; Skurray, R. Antimicrobial Resistance of Staphylococcus aureus: Genetic Basis. Microbiol. Rev. 1987, 51, 88–134.
- Vesterholm-Nielsen, M.; Larsen, M.O.; Olsen, J.E.; Aarestrup, F.M. Occurrence of the blaZ gene in penicillin resistant Staphylococcus aureus isolated from bovine mastitis in Denmark. Acta Vet. Scand. 1999, 40, 279–286.
- Sidhu, M.S.; Heir, E.; Leegaard, T.; Wiger, K.; Holck, A. Frequency of Disinfectant Resistance Genes and Genetic Linkage with β-Lactamase Transposon Tn552 among Clinical Staphylococci. Antimicrob. Agents Chemother. 2002, 46, 2797–2803.
- Murphy, E.; Novick, R.P. Physical Mapping of Staphylococcus aureus Penicillinase Plasmid pI524: Characterization of an Invertible Region. Mol. Gen. Genet. 1979, 175, 19–30.
- Sidhu, M.S.; Heir, E.; Sorum, H.; Holck, A. Genetic Linkage Between Resistance to Quaternary Ammonium Compounds and β-Lactam Antibiotics in Food-Related Staphylococcus spp. Microb. Drug Resist. 2001, 7, 363–371.
- Asheshov, E.H. The Genetics of Penicillinase Production in Staphylococcus aureus Strain PS80. J. Gen. Microbiol. 1969, 59, 289–301.
- Rowland, S.J.; Dyke, K.G. Tn552, a novel transposable element from Staphylococcus aureus. Mol. Microbiol. 1990, 4, 961–975.
- Wang, P.Z.; Projan, S.J.; Leason, K.R.; Novick, R.P. Translational Fusion with a Secretory Enzyme as an Indicator. J. Bacteriol. 1987, 169, 3082–3087.
- Miragaia, M. Factors Contributing to the Evolution of mecA-Mediated β-lactam Resistance in Staphylococci: Update and New Insights From Whole Genome Sequencing (WGS). Front. Microbiol. 2018, 9, 2723.
- Scherer, C.B.; Botoni, L.S.; Carvalho, A.U.; Keller, K.M.; Costa-Val, A.P. Ceftaroline resistance in Staphylococcus pseudintermedius gene mecA carriers. Pesqui. Vet. Bras. 2018, 38, 2233–2236.
- Long, S.W.; Olsen, R.J.; Mehta, S.C.; Palzkill, T.; Cernoch, P.L.; Perez, K.K.; Musick, W.L.; Rosato, A.E.; Musser, J.M. PBP2a Mutations Causing High-Level Ceftaroline Resistance in Clinical Methicillin-Resistant Staphylococcus aureus Isolates. Antimicrob. Agents Chemother. 2014, 58, 6668–6674.
- Hiramatsu, K. Molecular evolution of MRSA. Microbiol. Immunol. 1995, 39, 531–543.
- Deurenberg, R.H.; Stobberingh, E.E. The Molecular Evolution of Hospital- and Community-Associated Methicillin-Resistant Staphylococcus aureus. Curr. Mol. Med. 2009, 9, 100–115.
- Rasmussen, G.; Monecke, S.; Brus, O.; Ehricht, R.; Soderquist, B. Long Term Molecular Epidemiology of Methicillin-Susceptible Staphylococcus aureus Bacteremia Isolates in Sweden. PLoS ONE 2014, 9, e114276.
- Bruckner, R.; Matzura, H. Regulation of the inducible chloramphenicol acetyltransferase gene of the Staphylococcus aureus plasmid pUB112. EMBO J. 1985, 4, 2295–2300.
- Brückner, R.; Zyprian, E.; Matzura, H. Expression of a chloramphenicol-resistance determinant carried on hybrid plasmids in gram-positive and gram-negative bacteria. Gene 1984, 32, 151–160.
- Horinouchi, S.; Weisblum, B. Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J. Bacteriol. 1982, 150, 815–825.
- Shaw, W.V.; Brenner, D.G.; LeGrice, S.F.; Skinner, S.E.; Hawkins, A.R. Chloramphenicol acetyltransferase gene of staphylococcal plasmid pC221. Nucleotide sequence analysis and expression studies. Febs Lett. 1985, 179, 101–106.
- Macrina, F.L.; Archer, G.L. Conjugation and Broad Host Range Plasmids in Streptococci and Staphylococci. In Bacterial Conjugation; Clewell, D.B., Ed.; Springer: Boston, MA, USA, 1993; pp. 313–329.
- Koprivnjak, T.; Zhang, D.; Ernst, C.M.; Peschel, A.; Nauseef, W.M.; Weiss, J.P. Characterization of Staphylococcus aureus Cardiolipin Synthases 1 and 2 and Their Contribution to Accumulation of Cardiolipin in Stationary Phase and within Phagocytes. J. Bacteriol. 2011, 193, 4134–4142.
- Zhang, T.; Muraih, J.K.; Tishbi, N.; Herskowitz, J.; Victor, R.L.; Silverman, J.; Uwumarenogie, S.; Taylor, S.D.; Palmer, M.; Mintzer, E. Cardiolipin prevents membrane translocation and permeabilization by daptomycin. J. Biol. Chem. 2014, 289, 11584–11591.
- Jiang, J.H.; Bhuiyan, M.S.; Shen, H.H.; Cameron, D.R.; Rupasinghe, T.W.T.; Wu, C.M.; Le Brun, A.P.; Kostoulias, X.; Domene, C.; Fulcher, A.J.; et al. Antibiotic resistance and host immune evasion in Staphylococcus aureus mediated by a metabolic adaptation. Proc. Natl. Acad. Sci. USA 2019, 116, 3722–3727.
- Thitiananpakorn, K.; Aiba, Y.; Tan, X.E.; Watanabe, S.; Kiga, K.; Sato’o, Y.; Boonsiri, T.; Li, F.Y.; Sasahara, T.; Taki, Y.; et al. Association of mprF mutations with cross-resistance to daptomycin and vancomycin in methicillin-resistant Staphylococcus aureus (MRSA). Sci. Rep. 2020, 10, 16107.
- Chen, F.J.; Lauderdale, T.L.; Lee, C.H.; Hsu, Y.C.; Huang, I.W.; Hsu, P.C.; Yang, C.S. Effect of a Point Mutation in mprF on Susceptibility to Daptomycin, Vancomycin, and Oxacillin in an MRSA Clinical Strain. Front. Microbiol. 2018, 9, 1086.
- Mishra, N.N.; Yang, S.J.; Sawa, A.; Rubio, A.; Nast, C.C.; Yeaman, M.R.; Bayer, A.S. Analysis of Cell Membrane Characteristics of In Vitro-Selected Daptomycin-Resistant Strains of Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2009, 53, 2312–2318.
- Zuo, H.; Uehara, Y.; Lu, Y.J.; Sasaki, T.; Hiramatsu, K. Genetic and phenotypic diversity of methicillin-resistant Staphylococcus aureus among Japanese inpatients in the early 1980s. Sci. Rep. 2021, 11, 5447.
- Cui, L.Z.; Isii, T.; Fukuda, M.; Ochiai, T.; Neoh, H.M.; Camargo, I.L.B.D.; Watanabe, Y.; Shoji, M.; Hishinuma, T.; Hiramatsu, K. An RpoB Mutation Confers Dual Heteroresistance to Daptomycin and Vancomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 2010, 54, 5222–5233.
- Gao, W.; Cameron, D.R.; Davies, J.K.; Kostoulias, X.; Stepnell, J.; Tuck, K.L.; Yeaman, M.R.; Peleg, A.Y.; Stinear, T.P.; Howden, B.P. The RpoB H481Y rifampicin resistance mutation and an active stringent response reduce virulence and increase resistance to innate immune responses in Staphylococcus aureus. J. Infect. Dis. 2013, 207, 929–939.
- Howden, B.P.; McEvoy, C.R.E.; Allen, D.L.; Chua, K.; Gao, W.; Harrison, P.F.; Bell, J.; Coombs, G.; Bennett-Wood, V.; Porter, J.L.; et al. Evolution of Multidrug Resistance during Staphylococcus aureus Infection Involves Mutation of the Essential Two Component Regulator WalKR. PLoS Pathog. 2011, 7, e1002359.
- Poupel, O.; Moyat, M.; Groizeleau, J.; Antunes, L.C.S.; Gribaldo, S.; Msadek, T.; Dubrac, S. Transcriptional Analysis and Subcellular Protein Localization Reveal Specific Features of the Essential WalKR System in Staphylococcus aureus. PLoS ONE 2016, 11, e0151449.
- Dubrac, S.; Boneca, I.G.; Poupel, O.; Msadek, T. New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism and biofilm formation in Staphylococcus aureus. J. Bacteriol. 2007, 189, 8257–8269.
- Delaune, A.; Dubrac, S.; Blanchet, C.; Poupel, O.; Mader, U.; Hiron, A.; Leduc, A.; Fitting, C.; Nicolas, P.; Cavaillon, J.M.; et al. The WalKR System Controls Major Staphylococcal Virulence Genes and Is Involved in Triggering the Host Inflammatory Response. Infect. Immun. 2012, 80, 3438–3453.
- Mehta, S.; Cuirolo, A.X.; Plata, K.B.; Riosa, S.; Silverman, J.A.; Rubio, A.; Rosato, R.R.; Rosato, A.E. VraSR Two-Component Regulatory System Contributes to mprF-Mediated Decreased Susceptibility to Daptomycin in In Vivo-Selected Clinical Strains of Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2012, 56, 92–102.
- Gardete, S.; Wu, S.W.; Gill, S.; Tomasz, A. Role of VraSR in antibiotic resistance and antibiotic-induced stress response in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 3424–3434.
- Yin, S.H.; Daum, R.S.; Boyle-Vavra, S. VraSR Two-Component Regulatory System and Its Role in Induction of pbp2 and vraSR Expression by Cell Wall Antimicrobials in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 336–343.
- Kuroda, M.; Ohta, T.; Uchiyama, I.; Baba, T.; Yuzawa, H.; Kobayashi, I.; Cui, L.Z.; Oguchi, A.; Aoki, K.; Nagai, Y.; et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 2001, 357, 1225–1240.
- Kuroda, M.; Kuroda, H.; Oshima, T.; Takeuchi, F.; Mori, H.; Hiramatsu, K. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 2003, 49, 807–821.
- Courvalin, P. Vancomycin Resistance in Gram-Positive Cocci. Clin. Infect. Dis. 2006, 42 (Suppl. S1), S25–S34.
- Weigel, L.M.; Clewell, D.B.; Gill, S.R.; Clark, N.C.; McDougal, L.K.; Flannagan, S.E.; Kolonay, J.F.; Shetty, J.; Killgore, G.E.; Tenover, F.C. Genetic Analysis of a High-Level Vancomycin-Resistant Isolate of Staphylococcus aureus. Science 2003, 302, 1569–1571.
- Zhu, W.; Clark, N.C.; McDougal, L.K.; Hageman, J.; McDonald, L.C.; Patel, J.B. Vancomycin-resistant Staphylococcus aureus isolates associated with Inc18-like vanA plasmids in Michigan. Antimicrob. Agents Chemother. 2008, 52, 452–457.
- Périchon, B.; Courvalin, P. VanA-type vancomycin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2009, 53, 4580–4587.
- Malachowa, N.; DeLeo, F.R. Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010, 67, 3057–3071.
- Lannergard, J.; Norstrom, T.; Hughes, D. Genetic Determinants of Resistance to Fusidic Acid among Clinical Bacteremia Isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 2009, 53, 2059–2065.
- O’Neill, A.J.; Chopra, I. Molecular basis of fusB-mediated resistance to fusidic acid in Staphylococcus aureus. Mol. Microbiol. 2006, 59, 664–676.
- O’Brien, F.G.; Price, C.; Grubb, W.B.; Gustafson, J.E. Genetic characterization of the fusidic acid and cadmium resistance determinants of Staphylococcus aureus plasmid pUB101. J. Antimicrob. Chemother. 2002, 50, 313–321.
- O’Neill, A.J.; McLaws, F.; Kahlmeter, G.; Henriksen, A.S.; Chopra, I. Genetic basis of resistance to fusidic acid in staphylococci. Antimicrob. Agents Chemother. 2007, 51, 1737–1740.
- Kinnevey, P.M.; Shore, A.C.; Brennan, G.I.; Sullivan, D.J.; Ehricht, R.; Monecke, S.; Slickers, P.; Coleman, D.C. Emergence of Sequence Type 779 Methicillin-Resistant Staphylococcus aureus Harboring a Novel Pseudo Staphylococcal Cassette Chromosome mec (SCCmec)-SCC-SCCCRISPR Composite Element in Irish Hospitals. Antimicrob. Agents Chemother. 2013, 57, 524–531.
- Ender, M.; Berger-Bächi, B.; McCallum, N. Variability in SCCmecN1 spreading among injection drug users in Zurich, Switzerland. BMC Microbiol. 2007, 7, 62.
- Holden, M.T.; Feil, E.J.; Lindsay, J.A.; Peacock, S.J.; Day, N.P.; Enright, M.C.; Foster, T.J.; Moore, C.E.; Hurst, L.; Atkin, R.; et al. Complete genomes of two clinical Staphylococcus aureus strains: Evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. USA 2004, 101, 9786–9791.
- Lin, Y.-T.; Tsai, J.-C.; Chen, H.-J.; Hung, W.-C.; Hsueh, P.-R.; Teng, L.-J. A Novel Staphylococcal Cassette Chromosomal Element, SCCfusC, Carrying fusC and speG in Fusidic Acid-Resistant Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 1224–1227.
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. The Cfr rRNA Methyltransferase Confers Resistance to Phenicols, Lincosamides, Oxazolidinones, Pleuromutilins, and Streptogramin A Antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2500–2505.
- Morales, G.; Picazo, J.J.; Baos, E.; Candel, F.J.; Arribi, A.; Pelaez, B.; Andrade, R.; de la Torre, M.A.; Fereres, J.; Sanchez-Garcia, M. Resistance to Linezolid Is Mediated by the cfr Gene in the First Report of an Outbreak of Linezolid-Resistant Staphylococcus aureus. Clin. Infect. Dis. 2010, 50, 821–825.
- Kehrenberg, C.; Schwarz, S.; Jacobsen, L.; Hansen, L.H.; Vester, B. A new mechanism for chloramphenicol, florfenicol and clindamycin resistance: Methylation of 23S ribosomal RNA at A2503. Mol. Microbiol. 2005, 57, 1064–1073.
- Besier, S.; Ludwig, A.; Zander, J.; Brade, V.; Wichelhaus, T.A. Linezolid Resistance in Staphylococcus aureus: Gene Dosage Effect, Stability, Fitness Costs, and Cross-Resistances. Antimicrob. Agents Chemother. 2008, 52, 1570–1572.
- Toh, S.M.; Xiong, L.; Arias, C.A.; Villegas, M.V.; Lolans, K.; Quinn, J.; Mankin, A.S. Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol. Microbiol. 2007, 64, 1506–1514.
- Mendes, R.E.; Deshpande, L.M.; Bonilla, H.F.; Schwarz, S.; Huband, M.D.; Jones, R.N.; Quinn, J.P. Dissemination of a pSCFS3-Like cfr-Carrying Plasmid in Staphylococcus aureus and Staphylococcus epidermidis Clinical Isolates Recovered from Hospitals in Ohio. Antimicrob. Agents Chemother. 2013, 57, 2923–2928.
- Shore, A.C.; Brennan, O.M.; Ehricht, R.; Monecke, S.; Schwarz, S.; Slickers, P.; Coleman, D.C. Identification and characterization of the multidrug resistance gene cfr in a Panton-Valentine leukocidin-positive sequence type 8 methicillin-resistant Staphylococcus aureus IVa (USA300) isolate. Antimicrob. Agents Chemother. 2010, 54, 4978–4984.
- Shore, A.C.; Lazaris, A.; Kinnevey, P.M.; Brennan, O.M.; Brennan, G.I.; O’Connell, B.; Feßler, A.T.; Schwarz, S.; Coleman, D.C. First Report of cfr-Carrying Plasmids in the Pandemic Sequence Type 22 Methicillin-Resistant Staphylococcus aureus Staphylococcal Cassette Chromosome mec Type IV Clone. Antimicrob. Agents Chemother. 2016, 60, 3007–3015.
- Locke, J.B.; Rahawi, S.; Lamarre, J.; Mankin, A.S.; Shaw, K.J. Genetic Environment and Stability of cfr in Methicillin-Resistant Staphylococcus aureus CM05. Antimicrob. Agents Chemother. 2012, 56, 332–340.
- Zhu, Y.; Zhang, W.; Wang, C.; Liu, W.; Yang, Q.; Luan, T.; Wang, L.; Schwarz, S.; Liu, S. Identification of a novel optrA-harbouring transposon, Tn6823, in Staphylococcus aureus. J. Antimicrob. Chemother. 2020, 75, 3395–3397.
- Locke, J.B.; Hilgers, M.; Shaw, K.J. Novel Ribosomal Mutations in Staphylococcus aureus Strains Identified through Selection with the Oxazolidinones Linezolid and Torezolid (TR-700). Antimicrob. Agents Chemother. 2009, 53, 5265–5274.
- Lowy, F.D. Antimicrobial resistance: The example of Staphylococcus aureus. J. Clin. Investig. 2003, 111, 1265–1273.
- Saribas, Z.; Tunckanat, F.; Pinar, A. Prevalence of erm genes encoding macrolide-lincosamide-streptogramin (MLS) resistance among clinical isolates of Staphylococcus aureus in a Turkish university hospital. Clin. Microbiol. Infect. 2006, 12, 797–799.
- Leclercq, R.; Courvalin, P. Bacterial resistance to macrolide, lincosamide, and streptogramin antibiotics by target modification. Antimicrob. Agents Chemother. 1991, 35, 1267–1272.
- Schmitz, F.J.; Petridou, J.; Astfalk, N.; Scheuring, S.; Köhrer, K.; Verhoef, J.; Fluit, A.C.; Schwarz, S. Structural alterations in the translational attenuator of constitutively expressed erm(A) genes in Staphylococcus aureus. Antimicrob. Agents Chemother. 2001, 45, 1603–1604.
- Ito, T.; Katayama, Y.; Asada, K.; Mori, N.; Tsutsumimoto, K.; Tiensasitorn, C.; Hiramatsu, K. Structural comparison of three types of staphylococcal cassette chromosome mec integrated in the chromosome in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2001, 45, 1323–1336.
- Li, B.; Wendlandt, S.; Yao, J.; Liu, Y.; Zhang, Q.; Shi, Z.; Wei, J.; Shao, D.; Schwarz, S.; Wang, S.; et al. Detection and new genetic environment of the pleuromutilin-lincosamide-streptogramin A resistance gene lsa(E) in methicillin-resistant Staphylococcus aureus of swine origin. J. Antimicrob. Chemother. 2013, 68, 1251–1255.
- Sarrou, S.; Liakopoulos, A.; Tsoumani, K.; Sagri, E.; Mathiopoulos, K.D.; Tzouvelekis, L.S.; Miriagou, V.; Petinaki, E. Characterization of a Novel lsa(E)- and lnu(B)-Carrying Structure Located in the Chromosome of a Staphylococcus aureus Sequence Type 398 Strain. Antimicrob. Agents Chemother. 2016, 60, 1164–1166.
- Ji, X.; Krüger, H.; Wang, Y.; Feßler, A.T.; Wang, Y.; Schwarz, S.; Wu, C. Tn560, a Novel Tn554 Family Transposon from Porcine Methicillin-Resistant Staphylococcus aureus ST398, Carries a Multiresistance Gene Cluster Comprising a Novel spc Gene Variant and the Genes lsa(E) and lnu(B). Antimicrob. Agents Chemother. 2022, 66, e01947-21.
- Huang, J.; O’Toole, P.W.; Shen, W.; Amrine-Madsen, H.; Jiang, X.; Lobo, N.; Palmer, L.M.; Voelker, L.; Fan, F.; Gwynn, M.N.; et al. Novel chromosomally encoded multidrug efflux transporter MdeA in Staphylococcus aureus. Antimicrob. Agents Chemother. 2004, 48, 909–917.
- Matsuoka, M.; Endou, K.; Kobayashi, H.; Inoue, M.; Nakajima, Y. A plasmid that encodes three genes for resistance to macrolide antibiotics in Staphylococcus aureus. FEMS Microbiol. Lett. 1998, 167, 221–227.
- Udo, E.E.; Al-Sweih, N.; Noronha, B.C. A chromosomal location of the mupA gene in Staphylococcus aureus expressing high-level mupirocin resistance. J. Antimicrob. Chemother. 2003, 51, 1283–1286.
- Seah, C.; Alexander, D.C.; Louie, L.; Simor, A.; Low, D.E.; Longtin, J.; Melano, R.G. MupB, a New High-Level Mupirocin Resistance Mechanism in Staphylococcus aureus. Antimicrob. Agents Chemother. 2012, 56, 1916–1920.
- Woodford, N.; Watson, A.P.; Patel, S.; Jevon, M.; Waghorn, D.J.; Cookson, B.D. Heterogeneous location of the mupA high-level mupirocin resistance gene in Staphylococcus aureus. J. Med. Microbiol. 1998, 47, 829–835.
- Udo, E.E.; Jacob, L.E. Conjugative transfer of high-level mupirocin resistance and the mobilization of non-conjugative plasmids in Staphylococcus aureus. Microb. Drug Resist. 1998, 4, 185–193.
- Dyke, K.G.; Curnock, S.P.; Golding, M.; Noble, W.C. Cloning of the gene conferring resistance to mupirocin in Staphylococcus aureus. FEMS Microbiol. Lett. 1991, 61, 195–198.
- Goswami, C.; Fox, S.; Holden, M.; Leanord, A.; Evans, T.J. Genomic Analysis of Global Staphylococcus argenteus Strains Reveals Distinct Lineages With Differing Virulence and Antibiotic Resistance Gene Content. Front. Microbiol. 2021, 12, 795173.
- Etienne, J.; Gerbaud, G.; Courvalin, P.; Fleurette, J. Plasmid-mediated resistance to fosfomycin in Staphylococcus epidermidis. FEMS Microbiol. Lett. 1989, 52, 133–137.
- Fey, P.D.; Endres, J.L.; Yajjala, V.K.; Widhelm, T.J.; Boissy, R.J.; Bose, J.L.; Bayles, K.W. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 2013, 4, e00537-12.
- Fu, Z.; Liu, Y.; Chen, C.; Guo, Y.; Ma, Y.; Yang, Y.; Hu, F.; Xu, X.; Wang, M. Characterization of Fosfomycin Resistance Gene, fosB, in Methicillin-Resistant Staphylococcus aureus Isolates. PLoS ONE 2016, 11, e0154829.
- Zilhao, R.; Courvalin, P. Nucleotide sequence of the fosB gene conferring fosfomycin resistance in Staphylococcus epidermidis. FEMS Microbiol. Lett. 1990, 56, 267–272.
- Novick, R.P.; Christie, G.E.; Penadés, J.R. The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 2010, 8, 541–551.
- Sanfilippo, C.M.; Hesje, C.K.; Haas, W.; Morris, T.W. Topoisomerase Mutations That Are Associated with High-Level Resistance to Earlier Fluoroquinolones in Staphylococcus aureus Have Less Effect on the Antibacterial Activity of Besifloxacin. Chemotherapy 2011, 57, 363–371.
- Neyfakh, A.A.; Borsch, C.M.; Kaatz, G.W. Fluoroquinolone Resistance Protein NorA of Staphylococcus aureus Is a Multidrug Efflux Transporter. Antimicrob. Agents Chemother. 1993, 37, 128–129.
- Abdu, A.B.; Mirabeau, T.Y. Prevalence of qnr Genes among Multidrug Resistance Staphylococcus aureus from Clinical Isolates. J. Adv. Med. Med. Res. 2019, 30, 1–10.
- Haroche, J.; Allignet, J.; El Solh, N. Tn5406, a new staphylococcal transposon conferring resistance to streptogramin A and related compounds including dalfopristin. Antimicrob. Agents Chemother. 2002, 46, 2337–2343.
- Li, J.; Li, B.; Wendlandt, S.; Schwarz, S.; Wang, Y.; Wu, C.; Ma, Z.; Shen, J. Identification of a novel vga(E) gene variant that confers resistance to pleuromutilins, lincosamides and streptogramin A antibiotics in staphylococci of porcine origin. J. Antimicrob. Chemother. 2013, 69, 919–923.
- Lozano, C.; Aspiroz, C.; Rezusta, A.; Gómez-Sanz, E.; Simon, C.; Gómez, P.; Ortega, C.; Revillo, M.J.; Zarazaga, M.; Torres, C. Identification of novel vga(A)-carrying plasmids and a Tn5406-like transposon in meticillin-resistant Staphylococcus aureus and Staphylococcus epidermidis of human and animal origin. Int. J. Antimicrob. Agents 2012, 40, 306–312.
- Haroche, J.; Allignet, J.; Buchrieser, C.; El Solh, N. Characterization of a variant of vga(A) conferring resistance to streptogramin A and related compounds. Antimicrob. Agents Chemother. 2000, 44, 2271–2275.
- Donhofer, A.; Franckenberg, S.; Wickles, S.; Berninghausen, O.; Beckmann, R.; Wilson, D.N. Structural basis for TetM-mediated tetracycline resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 16900–16905.
- Lima, M.C.; de Barros, M.; Scatamburlo, T.M.; Polyeiro, R.C.; de Castro, L.K.; Guimaraes, S.H.S.; da Costa, S.L.; da Costa, M.M.; Moreira, M.A.S. Profiles of Staphyloccocus aureus isolated from goat persistent mastitis before and after treatment with enrofloxacin. BMC Microbiol. 2020, 20, 127.
- Emaneini, M.; Bigverdi, R.; Kalantar, D.; Soroush, S.; Jabalameli, F.; Noorazar Khoshgnab, B.; Asadollahi, P.; Taherikalani, M. Distribution of genes encoding tetracycline resistance and aminoglycoside modifying enzymes in Staphylococcus aureus strains isolated from a burn center. Ann. Burns Fire Disasters 2013, 26, 76–80.
- Guay, G.G.; Khan, S.A.; Rothstein, D.M. The tet(K) Gene of Plasmid pT181 of Staphylococcus aureus Encodes an Efflux Protein That Contains 14 Transmembrane Helices. Plasmid 1993, 30, 163–166.
- Jensen, S.O.; Lyon, B.R. Genetics of antimicrobial resistance in Staphylococcus aureus. Future Microbiol. 2009, 4, 565–582.
- Leroy, S.; Christieans, S.; Talon, R. Tetracycline Gene Transfer in Staphylococcus xylosus in situ During Sausage Fermentation. Front. Microbiol. 2019, 10, 392.
- Coque, T.M.; Singh, K.V.; Weinstock, G.M.; Murray, B.E. Characterization of Dihydrofolate Reductase Genes from Trimethoprim-Susceptible and Trimethoprim-Resistant Strains of Enterococcus faecalis. Antimicrob. Agents Chemother. 1999, 43, 141–147.
- Rouch, D.A.; Messerotti, L.J.; Loo, L.S.; Jackson, C.A.; Skurray, R.A. Trimethoprim resistance transposon Tn4003 from Staphylococcus aureus encodes genes for a dihydrofolate reductase and thymidylate synthetase flanked by three copies of IS257. Mol. Microbiol. 1989, 3, 161–175.
- Reeve, S.M.; Scocchera, E.W.; Narendran, G.D.; Keshipeddy, S.; Krucinska, J.; Hajian, B.; Ferreira, J.; Nailor, M.; Aeschlimann, J.; Wright, D.L.; et al. MRSA Isolates from United States Hospitals Carry dfrG and dfrK Resistance Genes and Succumb to Propargyl-Linked Antifolates. Cell Chem. Biol. 2016, 23, 1458–1467.
- Dale, G.E.; Broger, C.; DArcy, A.; Hartman, P.G.; DeHoogt, R.; Jolidon, S.; Kompis, I.; Labhardt, A.M.; Langen, H.; Locher, H.; et al. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J. Mol. Biol. 1997, 266, 23–30.
- Dale, G.E.; Broger, C.; Hartman, P.G.; Langen, H.; Page, M.G.P.; Then, R.L.; Stuber, D. Characterization of the Gene for the Chromosomal Dihydrofolate Reductase (DHFR) of Staphylococcus epidermidis ATCC 14990: The Origin of the Trimethoprim-Resistant S1 DHFR from Staphylococcus aureus? J. Bacteriol. 1995, 177, 2965–2970.
- Rodríguez-Martínez, J.M. Mechanisms of plasmid-mediated resistance to quinolones. Enferm. Infecc. Microbiol. Clin. 2005, 23, 25–31.
- Ulrich, N.; Vonberg, R.P.; Gastmeier, P. Outbreaks caused by vancomycin-resistant Enterococcus faecium in hematology and oncology departments: A systematic review. Heliyon 2017, 3, e00473.
- Willems, R.J.; Top, J.; van Santen, M.; Robinson, D.A.; Coque, T.M.; Baquero, F.; Grundmann, H.; Bonten, M.J. Global spread of vancomycin-resistant Enterococcus faecium from distinct nosocomial genetic complex. Emerg. Infect. Dis. 2005, 11, 821–828.
- Mahony, A.A.; Buultjens, A.H.; Ballard, S.A.; Grabsch, E.A.; Xie, S.; Seemann, T.; Stuart, R.L.; Kotsanas, D.; Cheng, A.; Heffernan, H.; et al. Vancomycin-resistant Enterococcus faecium sequence type 796—Rapid international dissemination of a new epidemic clone. Antimicrob. Resist. Infect. Control 2018, 7, 44.
- Wassilew, N.; Seth-Smith, H.M.; Rolli, E.; Fietze, Y.; Casanova, C.; Führer, U.; Egli, A.; Marschall, J.; Buetti, N. Outbreak of vancomycin-resistant Enterococcus faecium clone ST796, Switzerland, December 2017 to April 2018. Eurosurveillance 2018, 23, 1800351.
- Masalha, M.; Borovok, I.; Schreiber, R.; Aharonowitz, Y.; Cohen, G. Analysis of Transcription of the Staphylococcus aureus Aerobic Class Ib and Anaerobic Class III Ribonucleotide Reductase Genes in Response to Oxygen. J. Bacteriol. 2001, 183, 7260–7272.
- Parlet, C.P.; Brown, M.M.; Horswill, A.R. Commensal Staphylococci Influence Staphylococcus aureus Skin Colonization and Disease. Trends Microbiol. 2019, 27, 497–507.
- Uhlemann, A.-C.; Otto, M.; Lowy, F.D.; DeLeo, F.R. Evolution of community- and healthcare-associated methicillin-resistant Staphylococcus aureus. Infect. Genet. Evol. 2014, 21, 563–574.
- McNamee, P.T.; Smyth, J.A. Bacterial chondronecrosis with osteomyelitis (‘femoral head necrosis’) of broiler chickens: A review. Avian. Pathol. 2000, 29, 253–270.
- Peton, V.; Le Loir, Y. Staphylococcus aureus in veterinary medicine. Infect. Genet. Evol. 2014, 21, 602–615.
- Sakr, A.; Bregeon, F.; Mege, J.L.; Rolain, J.M.; Blin, O. Staphylococcus aureus Nasal Colonization: An Update on Mechanisms, Epidemiology, Risk Factors, and Subsequent Infections. Front. Microbiol. 2018, 9, 2419.
- Aires de Sousa, M.; de Lencastre, H. Bridges from hospitals to the laboratory: Genetic portraits of methicillin-resistant Staphylococcus aureus clones. FEMS Immunol. Med. Microbiol. 2004, 40, 101–111.
- Bien, J.; Sokolova, O.; Bozko, P. Characterization of Virulence Factors of Staphylococcus aureus: Novel Function of Known Virulence Factors That Are Implicated in Activation of Airway Epithelial Proinflammatory Response. J. Pathog. 2011, 2011, 601905.
- Oogai, Y.; Matsuo, M.; Hashimoto, M.; Kato, F.; Sugai, M.; Komatsuzawa, H. Expression of virulence factors by Staphylococcus aureus grown in serum. Appl. Environ. Microbiol. 2011, 77, 8097–8105.
- Silversides, J.A.; Lappin, E.; Ferguson, A.J. Staphylococcal Toxic Shock Syndrome: Mechanisms and Management. Curr. Infect. Dis. Rep. 2010, 12, 392–400.
- McGuinness, W.A.; Malachowa, N.; DeLeo, F.R. Vancomycin Resistance in Staphylococcus aureus. Yale J. Biol. Med. 2017, 90, 269–281.
- Peng, H.; Liu, D.; Ma, Y.; Gao, W. Comparison of community- and healthcare-associated methicillin-resistant Staphylococcus aureus isolates at a Chinese tertiary hospital, 2012–2017. Sci. Rep. 2018, 8, 17916.
- Xie, X.; Bao, Y.; Ouyang, N.; Dai, X.; Pan, K.; Chen, B.; Deng, Y.; Wu, X.; Xu, F.; Li, H.; et al. Molecular epidemiology and characteristic of virulence gene of community-acquired and hospital-acquired methicillin-resistant Staphylococcus aureus isolates in Sun Yat-sen Memorial hospital, Guangzhou, Southern China. BMC Infect. Dis. 2016, 16, 339.
- Figueiredo, A.M.; Ferreira, F.A. The multifaceted resources and microevolution of the successful human and animal pathogen methicillin-resistant Staphylococcus aureus. Mem. Inst. Oswaldo Cruz 2014, 109, 265–278.
- Figueiredo, A.M.S. What is behind the epidemiological difference between community-acquired and health-care associated methicillin-resistant Staphylococcus aureus? Virulence 2017, 8, 640–642.
- Naimi, T.S.; LeDell, K.H.; Como-Sabetti, K.; Borchardt, S.M.; Boxrud, D.J.; Etienne, J.; Johnson, S.K.; Vandenesch, F.; Fridkin, S.; O’Boyle, C.; et al. Comparison of Community- and Health Care–Associated Methicillin-Resistant Staphylococcus aureus Infection. JAMA 2003, 290, 2976–2984.
- Otto, M. Community-associated MRSA: What makes them special? Int. J. Med. Microbiol. 2013, 303, 324–330.
- Bloomfield, L.E.; Coombs, G.W.; Tempone, S.; Armstrong, P.K. Marked increase in community-associated methicillin-resistant Staphylococcus aureus infections, Western Australia, 2004-2018. Epidemiol. Infect. 2020, 148, e153.
- Agostino, J.W.; Ferguson, J.K.; Eastwood, K.; Kirk, M.D. The increasing importance of community-acquired methicillin-resistant Staphylococcus aureus infections. Med. J. Aust. 2017, 207, 388–393.
- Petersen, A.; Larssen, K.W.; Gran, F.W.; Enger, H.; Hæggman, S.; Mäkitalo, B.; Haraldsson, G.; Lindholm, L.; Vuopio, J.; Henius, A.E.; et al. Increasing Incidences and Clonal Diversity of Methicillin-Resistant Staphylococcus aureus in the Nordic Countries—Results From the Nordic MRSA Surveillance. Front. Microbiol. 2021, 12, 668900.
- Junnila, J.; Hirvioja, T.; Rintala, E.; Auranen, K.; Rantakokko-Jalava, K.; Silvola, J.; Lindholm, L.; Gröndahl-Yli-Hannuksela, K.; Marttila, H.; Vuopio, J. Changing epidemiology of methicillin-resistant Staphylococcus aureus in a low endemicity area—New challenges for MRSA control. Eur. J. Clin. Microbiol. 2020, 39, 2299–2307.
- Cameron, J.K.; Hall, L.; Tong, S.Y.C.; Paterson, D.L.; Halton, K. Incidence of community onset MRSA in Australia: Least reported where it is Most prevalent. Antimicrob. Resist. Infect. Control 2019, 8, 33.
- Macmorran, E.; Harch, S.; Athan, E.; Lane, S.; Tong, S.; Crawford, L.; Krishnaswamy, S.; Hewagama, S. The rise of methicillin resistant Staphylococcus aureus: Now the dominant cause of skin and soft tissue infection in Central Australia. Epidemiol. Infect. 2017, 145, 2817–2826.
- Baines, S.L.; Holt, K.E.; Schultz, M.B.; Seemann, T.; Howden, B.O.; Jensen, S.O.; Hal, S.J.v.; Coombs, G.W.; Firth, N.; Powell, D.R.; et al. Convergent Adaptation in the Dominant Global Hospital Clone ST239 of Methicillin-Resistant Staphylococcus aureus. mBio 2015, 6, e00080-15.
- Wang, B.; Xu, Y.; Zhao, H.; Wang, X.; Rao, L.; Guo, Y.; Yi, X.; Hu, L.; Chen, S.; Han, L.; et al. Methicillin-resistant Staphylococcus aureus in China: A multicentre longitudinal study and whole-genome sequencing. Emerg. Microbes Infect. 2022, 11, 532–542.
- Chen, H.; Yin, Y.; van Dorp, L.; Shaw, L.P.; Gao, H.; Acman, M.; Yuan, J.; Chen, F.; Sun, S.; Wang, X.; et al. Drivers of methicillin-resistant Staphylococcus aureus (MRSA) lineage replacement in China. Genome Med. 2021, 13, 171.
- Aires-de-Sousa, M. Methicillin-resistant Staphylococcus aureus among animals: Current overview. Clin. Microbiol. Infect. 2017, 23, 373–380.
- Lin, Y.; Barker, E.; Kislow, J.; Kaldhone, P.; Stemper, M.E.; Pantrangi, M.; Moore, F.M.; Hall, M.; Fritsche, T.R.; Novicki, T.; et al. Evidence of multiple virulence subtypes in nosocomial and community-associated MRSA genotypes in companion animals from the upper midwestern and northeastern United States. Clin. Med. Res. 2011, 9, 7–16.
- DeLeo, F.R.; Chambers, H.F. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J. Clin. Investig. 2009, 119, 2464–2474.
- Nichol, K.A.; Adam, H.J.; Golding, G.R.; Lagacé-Wiens, P.R.S.; Karlowsky, J.A.; Hoban, D.J.; Zhanel, G.G. Characterization of MRSA in Canada from 2007 to 2016. J. Antimicrob. Chemother. 2019, 74, iv55–iv63.
- Reyes, J.; Carvajal, L.P.; Rios, R.; Echeverri, A.; Rincon, S.; Munita, J.M.; Tran, T.; Panesso, D.; Arias, C.; Diaz, L. 1221. Genetic Characteristics of Healthcare-Associated Methicillin-Resistant Staphylococcus aureus (HA-MRSA) Belonging to Clonal Complex 5 (CC5) in Latin-America. Open Forum Infect. Dis. 2018, 5, S370.
- Arias, C.A.; Reyes, J.; Carvajal, L.P.; Rincon, S.; Diaz, L.; Panesso, D.; Ibarra, G.; Rios, R.; Munita, J.M.; Salles, M.J.; et al. A Prospective Cohort Multicenter Study of Molecular Epidemiology and Phylogenomics of Staphylococcus aureus Bacteremia in Nine Latin American Countries. Antimicrob. Agents Chemother. 2017, 61, e00816-17.
- Coombs, G.W.; Daley, D.A.; Yee, N.W.T.; Shoby, P.; Mowlaboccus, S. Australian Group on Antimicrobial Resistance (AGAR) Australian Staphylococcus aureus Sepsis Outcome Programme (ASSOP) Annual Report 2020. Commun. Dis. Intell. 2022, 46, 2018.
- Abdulgader, S.M.; Shittu, A.O.; Nicol, M.P.; Kaba, M. Molecular epidemiology of Methicillin-resistant Staphylococcus aureus in Africa: A systematic review. Front. Microbiol. 2015, 6, 348.
- Junie, L.M.; Jeican, I.I.; Matroş, L.; Pandrea, S.L. Molecular epidemiology of the community-associated methicillin-resistant Staphylococcus aureus clones: A synthetic review. Clujul Med. 2018, 91, 7–11.
- Gosbell, I.B. Epidemiology, clinical features and management of infections due to community methicillin-resistant Staphylococcus aureus (cMRSA). Intern. Med. J. 2005, 35 (Suppl. S2), S120–S135.
- O’Brien, F.G.; Pearman, J.W.; Gracey, M.; Riley, T.V.; Grubb, W.B. Community strain of methicillin-resistant Staphylococcus aureus involved in a hospital outbreak. J. Clin. Microbiol. 1999, 37, 2858–2862.
- Preeja, P.P.; Kumar, S.H.; Shetty, V. Prevalence and Characterization of Methicillin-Resistant Staphylococcus aureus from Community- and Hospital-Associated Infections: A Tertiary Care Center Study. Antibiotics 2021, 10, 197.
- Udo, E.E.; Aly, N.Y.A.; Sarkhoo, E.; Al-Sawan, R.; Al-Asar, A.M. Detection and characterization of an ST97-SCCmec-V community-associated meticillin-resistant Staphylococcus aureus clone in a neonatal intensive care unit and special care baby unit. J. Med. Microbiol. 2011, 60, 600–604.
- Joo, E.J.; Chung, D.R.; Ha, Y.E.; Park, S.Y.; Kang, S.J.; Kim, S.H.; Kang, C.I.; Peck, K.R.; Lee, N.Y.; Ko, K.S.; et al. Community-associated Panton-Valentine leukocidin-negative meticillin-resistant Staphylococcus aureus clone (ST72-MRSA-IV) causing healthcare-associated pneumonia and surgical site infection in Korea. J. Hosp. Infect. 2012, 81, 149–155.
- Park, S.H.; Park, C.; Yoo, J.H.; Choi, S.M.; Choi, J.H.; Shin, H.H.; Lee, D.G.; Lee, S.; Kim, J.; Choi, S.E.; et al. Emergence of community-associated methicillin-resistant Staphylococcus aureus strains as a cause of healthcare-associated bloodstream infections in Korea. Infect. Control Hosp. Epidemiol. 2009, 30, 146–155.
- Valsesia, G.; Rossi, M.; Bertschy, S.; Pfyffer, G.E. Emergence of SCCmec type IV and SCCmec type V methicillin-resistant Staphylococcus aureus containing the Panton-Valentine leukocidin genes in a large academic teaching hospital in central Switzerland: External invaders or persisting circulators? J. Clin. Microbiol. 2010, 48, 720–727.
- Sonnevend, Á.; Blair, I.; Alkaabi, M.; Jumaa, P.; Al Haj, M.; Ghazawi, A.; Akawi, N.; Jouhar, F.S.; Hamadeh, M.B.; Pál, T. Change in meticillin-resistant Staphylococcus aureus clones at a tertiary care hospital in the United Arab Emirates over a 5-year period. J. Clin. Pathol. 2012, 65, 178–182.
- Gould, I.M.; Girvan, E.K.; Browning, R.A.; Mackenzie, F.M.; Edwards, G.F.S. Report of a hospital neonatal unit outbreak of community-associated methicillin-resistant Staphylococcus aureus. Epidemiol. Infect. 2009, 137, 1242–1248.
- Maree, C.L.; Daum, R.S.; Boyle-Vavra, S.; Matayoshi, K.; Miller, L.G. Community-associated methicillin-resistant Staphylococcus aureus isolates causing healthcare-associated infections. Emerg. Infect. Dis. 2007, 13, 236–242.
- Patel, M.; Hoesley, C.J.; Moser, S.A.; Stamm, A.M.; Baddley, J.W.; Waites, K.B. Dissemination of community-associated methicillin-resistant Staphylococcus aureus in a tertiary care hospital. Antibiotics 2008, 101, 40–45.
- David, M.Z.; Cadilla, A.; Boyle-Vavra, S.; Daum, R.S. Replacement of HA-MRSA by CA-MRSA infections at an academic medical center in the midwestern United States, 2004–2005 to 2008. PLoS ONE 2014, 9, e92760.
- Baldan, R.; Testa, F.; Lorè, N.I.; Bragonzi, A.; Cichero, P.; Ossi, C.; Biancardi, A.; Nizzero, P.; Moro, M.; Cirillo, D.M. Factors contributing to epidemic MRSA clones replacement in a hospital setting. PLoS ONE 2012, 7, e43153.
- Planet, P.J. Life After USA300: The Rise and Fall of a Superbug. J. Infect. Dis. 2017, 215, S71–S77.
- Chambers, H.F.; Deleo, F.R. Waves of Resistance: Staphylococcus aureus in the Antibiotic Era. Nat. Rev. Microbiol. 2009, 7, 629–641.
- Lawal, O.U.; Ayobami, O.; Abouelfetouh, A.; Mourabit, N.; Kaba, M.; Egyir, B.; Abdulgader, S.M.; Shittu, A.O. A 6-Year Update on the Diversity of Methicillin-Resistant Staphylococcus aureus Clones in Africa: A Systematic Review. Front. Microbiol. 2022, 13, 860436.
- Hiramatsu, K.; Hanaki, H.; Ino, T.; Yabuta, K.; Oguri, T.; Tenover, F.C. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 1997, 40, 135–136.
- Ohlsen, K. Novel antibiotics for the treatment of Staphylococcus aureus. Expert Rev. Clin. Pharmacol. 2009, 2, 661–672.
- Kluytmans, J.; vanBelkum, A.; Verbrugh, H. Nasal Carriage of Staphylococcus aureus: Epidemiology, Underlying Mechanisms, and Associated Risks. Clin. Microbiol. Rev. 1997, 10, 505–520.
- Grundmann, H.; Aires-De-Sousa, M.; Boyce, J.; Tiemersma, E. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet 2006, 368, 874–885.
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655.
- Kourtis, A.P.; Hatfield, K.; Baggs, J.; Mu, Y.; See, I.; Epson, E.; Nadle, J.; Kainer, M.A.; Dumyati, G.; Petit, S.; et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections—United States. MMWR Morb. Mortal. Wkly Rep. 2019, 68, 214–219.
- World Health Organisation. Global Priority List of Antibiotic Resistant Bacteria; World Health Organisation: Geneva, Switzerland, 2017; p. 7.
- Rubinstein, E.; Keynan, Y. Vancomycin revisited—60 years later. Front. Public Health 2014, 2, 217.
- Geraci, J.E.; Heilman, F.R.; Nichols, D.R.; Wellman, W.E.; Ross, G.T. Some laboratory and clinical experiences with a new antibiotic, vancomycin. Proc. Staff Meet. Mayo Clin. 1956, 48, 809–810.
- Geraci, J.E.; Heilman, F.R.; Nichols, D.R.; Wellman, W.E. Antibiotic therapy of bacterial endocarditis. VII. Vancomycin for acute micrococcal endocarditis; preliminary report. Proc. Staff Meet. Mayo Clin. 1958, 33, 172–181.
- Filippone, E.J.; Kraft, W.K.; Farber, J.L. The Nephrotoxicity of Vancomycin. Clin. Pharmacol. Ther. 2017, 102, 459–469.
- Hazlewood, K.A.; Brouse, S.D.; Pitcher, W.D.; Hall, R.G. Vancomycin-associated nephrotoxicity: Grave concern or death by character assassination? Am. J. Med. 2010, 123, e181–e187.
- Cong, Y.; Yang, S.; Rao, X. Vancomycin resistant Staphylococcus aureus infections: A review of case updating and clinical features. J. Adv. Res. 2020, 21, 169–176.
- Brown, N.M.; Goodman, A.L.; Horner, C.; Jenkins, A.; Brown, E.M. Treatment of methicillin-resistant Staphylococcus aureus (MRSA): Updated guidelines from the UK. JAC Antimicrob. Resist. 2021, 3, dlaa114.
- Choo, E.J.; Chambers, H.F. Treatment of Methicillin-Resistant Staphylococcus aureus Bacteremia. Infect. Chemother. 2016, 48, 267–273.
- Vemula, P.K.; Campbell, N.R.; Zhao, F.; Xu, B.; John, G.; Karp, J.M. 4.421—Self-Assembled Prodrugs. In Comprehensive Biomaterials; Ducheyne, P., Ed.; Elsevier: Oxford, UK, 2011; pp. 339–355.
- Patel, S.; Preuss, C.V.; Bernice, F. Vancomycin; StatPearls: Treasure Island, FL, USA, 2022.
- Chang, J.D.; Foster, E.E.; Wallace, A.G.; Kim, S.J. Peptidoglycan O-acetylation increases in response to vancomycin treatment in vancomycin-resistant Enterococcus faecalis. Sci. Rep. 2017, 7, 46500.
- Meziane-Cherif, D.; Stogios, P.J.; Evdokimova, E.; Savchenko, A.; Courvalin, P. Structural basis for the evolution of vancomycin resistance D,D-peptidases. Proc. Natl. Acad. Sci. USA 2014, 111, 5872–5877.
- Zawadzka-Skomial, J.; Markiewicz, Z.; Nguyen-Disteche, M.; Devreese, B.; Frere, J.M.; Terrak, M. Characterization of the bifunctional glycosyltransferase/acyltransferase penicillin-binding protein 4 of Listeria monocytogenes. J. Bacteriol. 2006, 188, 1875–1881.
- Hu, Q.W.; Peng, H.G.; Rao, X.C. Molecular Events for Promotion of Vancomycin Resistance in Vancomycin Intermediate Staphylococcus aureus. Front. Microbiol. 2016, 7, 1601.
- Wang, F.; Zhou, H.Y.; Olademehin, O.P.; Kim, S.J.; Tao, P. Insights into Key Interactions between Vancomycin and Bacterial Cell Wall Structures. ACS Omega 2018, 3, 37–45.
- Sinha Roy, R.; Yang, P.; Kodali, S.; Xiong, Y.; Kim, R.M.; Griffin, P.R.; Onishi, H.R.; Kohler, J.; Silver, L.L.; Chapman, K. Direct interaction of a vancomycin derivative with bacterial enzymes involved in cell wall biosynthesis. Chem. Biol. 2001, 8, 1095–1106.
- World Health Organisation. Selection and Use of Essential Medicines: Report of the WHO Expert Committee on Selection and Use of Essential Medicines, 2019 (Including the 21st WHO Model List of Essential Medicines and the 7th WHO Model List of Essential Medicines for Children); Report No.: 0512-3054; World Health Organisation: Geneva, Switzerland, 2019; pp. 1–639.
- Uttley, A.H.C.; Collins, C.H.; Naidoo, J.; George, R.C. Vancomycin-Resistant Enterococci. Lancet 1988, 1, 57–58.
- Clinical Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, 31st ed.; CLSI supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2021; Volume 41.
- The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters, 12th ed.; The European Committee on Antimicrobial Susceptibility Testing: Växjö, Sweden, 2022.
- Bugg, T.D.H.; Wright, G.D.; Dutkamalen, S.; Arthur, M.; Courvalin, P.; Walsh, C.T. Molecular Basis for Vancomycin Resistance in Enterococcus faecium BM4147: Biosynthesis of a Depsipeptide Peptidoglycan Precursor by Vancomycin Resistance Proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415.
- Stogios, P.J.; Savchenko, A. Molecular mechanisms of vancomycin resistance. Protein Sci. 2020, 29, 654–669.
- Billot-Klein, D.; Blanot, D.; Gutmann, L.; van Heijenoort, J. Association constants for the binding of vancomycin and teicoplanin to N-acetyl-D-alanyl-D-alanine and N-acetyl-D-alanyl-D-serine. Biochem. J. 1994, 304 Pt 3, 1021–1022.
- Arthur, M.; Quintiliani, R., Jr. Regulation of VanA- and VanB-Type Glycopeptide Resistance in Enterococci. Antimicrob. Agents Chemother. 2001, 45, 375–381.
- Marshall, C.G.; Zolli, M.; Wright, G.D. Molecular Mechanism of VanHst, an α-Ketoacid Dehydrogenase Required for Glycopeptide Antibiotic Resistance from a Glycopeptide Producing Organism. Biochemistry 1999, 38, 8485–8491.
- Arthur, M.; Molinas, C.; Courvalin, P. The VanS-VanR Two-Component Regulatory System Controls Synthesis of Depsipeptide Peptidoglycan Precursorsin Enterococcus faecium BM4147. J. Bacteriol. 1992, 174, 2582–2591.
- Wu, Z.; Wright, G.D.; Walsh, C.T. Overexpression, purification, and characterization of VanX, a D-, D-dipeptidase which is essential for vancomycin resistance in Enterococcus faecium BM4147. Biochemistry 1995, 34, 2455–2463.
- Arthur, M.; Depardieu, F.; Snaith, H.A.; Reynolds, P.E.; Courvalin, P. Contribution of VanY D,D-carboxypeptidase to glycopeptide resistance in Enterococcus faecalis by hydrolysis of peptidoglycan precursors. Antimicrob. Agents Chemother. 1994, 38, 1899–1903.
- Arthur, M.; Depardieu, F.; Cabanie, L.; Reynolds, P.; Courvalin, P. Requirement of the VanY and VanX D,D-peptidases for glycopeptide resistance in enterococci. Mol. Microbiol. 1998, 30, 819–830.
- Wright, G.D.; Molinas, C.; Arthur, M.; Courvalin, P.; Walsh, C.T. Characterization of vanY, a DD-carboxypeptidase from vancomycin-resistant Enterococcus faecium BM4147. Antimicrob. Agents Chemother. 1992, 36, 1514–1518.
- Smith, J.D.; Kumarasiri, M.; Zhang, W.; Hesek, D.; Lee, M.; Toth, M.; Vakulenko, S.; Fisher, J.F.; Mobashery, S.; Chen, Y. Structural analysis of the role of Pseudomonas aeruginosa penicillin-binding protein 5 in β-lactam resistance. Antimicrob. Agents Chemother. 2013, 57, 3137–3146.
- Peters, K.; Kannan, S.; Rao, V.A.; Biboy, J.; Vollmer, D.; Erickson, S.W.; Lewis, R.J.; Young, K.D.; Vollmer, W. The Redundancy of Peptidoglycan Carboxypeptidases Ensures Robust Cell Shape Maintenance in Escherichia coli. mBio 2016, 7, e00819-16.
- Arthur, M.; Depardieu, F.; Molinas, C.; Reynolds, P.; Courvalin, P. The vanZ gene of Tn1546 from Enterococcus faecium BM4147 confers resistance to teicoplanin. Gene 1995, 154, 87–92.
- Arthur, M.; Depardieu, F.; Reynolds, P.; Courvalin, P. Quantitative analysis of the metabolism of soluble cytoplasmic peptidoglycan precursors of glycopeptide-resistant enterococci. Mol. Microbiol. 1996, 21, 33–44.
- Lebreton, F.; Depardieu, F.; Bourdon, N.; Fines-Guyon, M.; Berger, P.; Camiade, S.; Leclercq, R.; Courvalin, P.; Cattoir, V. D-Ala-D-Ser VanN-type transferable vancomycin resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 2011, 55, 4606–4612.
- Boyd, D.A.; Willey, B.M.; Fawcett, D.; Gillani, N.; Mulvey, M.R. Molecular Characterization of Enterococcus faecalis N06-0364 with Low-Level Vancomycin Resistance Harboring a Novel D-Ala-D-Ser Gene Cluster, vanL. Antimicrob. Agents Chemother. 2008, 52, 2667–2672.
- Arias, C.A.; Weisner, J.; Blackburn, J.M.; Reynolds, P.E. Serine and alanine racemase activities of VanT: A protein necessary for vancomycin resistance in Enterococcus gallinarum BM4174. Microbiology 2000, 146 Pt 7, 1727–1734.
- Arias, C.A.; Martin-Martinez, M.; Blundell, T.L.; Arthur, M.; Courvalin, P.; Reynolds, P.E. Characterization and modelling of VanT: A novel, membrane-bound, serine racemase from vancomycin-resistant Enterococcus gallinarum BM4174. Mol. Microbiol. 1999, 31, 1653–1664.
- Arias, C.A.; Courvalin, P.; Reynolds, P.E. vanC Cluster of Vancomycin-Resistant Enterococcus gallinarum BM4174. Antimicrob. Agents Chemother. 2000, 44, 1660–1666.
- Abadía Patiño, L.; Courvalin, P.; Perichon, B. vanE gene cluster of vancomycin-resistant Enterococcus faecalis BM4405. J. Bacteriol. 2002, 184, 6457–6464.
- Depardieu, F.; Bonora, M.G.; Reynolds, P.E.; Courvalin, P. The vanG glycopeptide resistance operon from Enterococcus faecalis revisited. Mol. Microbiol. 2003, 50, 931–948.
- Arias, C.A.; Pena, J.; Panesso, D.; Reynolds, P. Role of the transmembrane domain of the VanT serine racemase in resistance to vancomycin in Enterococcus gallinarum BM4174. J. Antimicrob. Chemother. 2003, 51, 557–564.
- Meziane-Cherif, D.; Stogios, P.J.; Evdokimova, E.; Egorova, O.; Savchenko, A.; Courvalin, P. Structural and Functional Adaptation of Vancomycin Resistance VanT Serine Racemases. mBio 2015, 6, e00806.
- Espaillat, A.; Carrasco-López, C.; Bernardo-García, N.; Pietrosemoli, N.; Otero, L.H.; Álvarez, L.; de Pedro, M.A.; Pazos, F.; Davis, B.M.; Waldor, M.K.; et al. Structural basis for the broad specificity of a new family of amino-acid racemases. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 79–90.
- Wu, H.M.; Kuan, Y.C.; Chu, C.H.; Hsu, W.H.; Wang, W.C. Crystal structures of lysine-preferred racemases, the non-antibiotic selectable markers for transgenic plants. PLoS ONE 2012, 7, e48301.
- Podmore, A.H.; Reynolds, P.E. Purification and characterization of VanXYc, a D,D-dipeptidase/D,D-carboxypeptidase in vancomycin-resistant Enterococcus gallinarum BM4174. Eur. J. Biochem. 2002, 269, 2740–2746.
- Reynolds, P.E.; Arias, C.A.; Courvalin, P. Gene vanXYc encodes D,D -dipeptidase (VanX) and D,D-carboxypeptidase (VanY) activities in vancomycin-resistant Enterococcus gallinarum BM4174. Mol. Microbiol. 1999, 34, 341–349.
- Ahmed, M.O.; Baptiste, K.E. Vancomycin-Resistant Enterococci: A Review of Antimicrobial Resistance Mechanisms and Perspectives of Human and Animal Health. Microb. Drug Resist. 2018, 24, 590–606.
- Wu, Q.; Sabokroo, N.; Wang, Y.; Hashemian, M.; Karamollahi, S.; Kouhsari, E. Systematic review and meta-analysis of the epidemiology of vancomycin-resistance Staphylococcus aureus isolates. Antimicrob. Resist. Infect. Control 2021, 10, 101.
- Jacoby, G.A. Transmissible Antibiotic Resistance. In Antimicrobial Resistance in the 21st Century; Fong, I.W., Shlaes, D., Drlica, K., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 341–381.
- Hughes, D. Exploiting genomics, genetics and chemistry to combat antibiotic resistance. Nat. Rev. Genet. 2003, 4, 432–441.
- Centers for Disease Control and Prevention. Healthcare-associated Infections—VRE and the Clinical Laboratory. 2010. Available online: https://www.cdc.gov/hai/settings/lab/vreclinical-laboratory.html (accessed on 12 August 2022).
- Sahm, D.F.; Free, L.; Handwerger, S. Inducible and constitutive expression of vanC-1-encoded resistance to vancomycin in Enterococcus gallinarum. Antimicrob. Agents Chemother. 1995, 39, 1480–1484.
- Reynolds, P.E.; Snaith, H.A.; Maguire, A.J.; Dutka-Malen, S.; Courvalin, P. Analysis of peptidoglycan precursors in vancomycin-resistant Enterococcus gallinarum BM4174. Biochem. J. 1994, 301 Pt 1, 5–8.
- Reynolds, P.E.; Courvalin, P. Vancomycin Resistance in Enterococci Due to Synthesis of Precursors Terminating in D-Alanyl-D-Serine. Antimicrob. Agents Chemother. 2005, 49, 21–25.
- Naser, S.M.; Vancanneyt, M.; Hoste, B.; Snauwaert, C.; Vandemeulebroecke, K.; Swings, J. Reclassification of Enterococcus flavescens Pompei et al. 1992 as a later synonym of Enterococcus casseliflavus (ex Vaughan et al. 1979) Collins et al. 1984 and Enterococcus saccharominimus Vancanneyt et al. 2004 as a later synonym of Enterococcus italicus Fortina et al. 2004. Int. J. Syst. Evol. Microbiol. 2006, 56, 413–416.
- Dutta, I.; Reynolds, P.E. Biochemical and Genetic Characterization of the vanC-2 Vancomycin Resistance Gene Cluster of Enterococcus casseliflavus ATCC 25788. Antimicrob. Agents Chemother. 2002, 46, 3125–3132.
- Fines, M.; Perichon, B.; Reynolds, P.; Sahm, D.F.; Courvalin, P. VanE, a New Type of Acquired Glycopeptide Resistance in Enterococcus faecalis BM4405. Antimicrob. Agents Chemother. 1999, 43, 2161–2164.
- McKessar, S.J.; Berry, A.M.; Bell, J.M.; Turnidge, J.D.; Paton, J.C. Genetic characterization of vanG, a novel vancomycin resistance locus of Enterococcus faecalis. Antimicrob. Agents Chemother. 2000, 44, 3224–3228.
- Hiramatsu, K.; Aritaka, N.; Hanaki, H.; Kawasaki, S.; Hosoda, Y.; Hori, S.; Fukuchi, Y.; Kobayashi, I. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 1997, 350, 1670–1673.
- Hiramatsu, K.; Kayayama, Y.; Matsuo, M.; Aiba, Y.; Saito, M.; Hishinuma, T.; Iwamoto, A. Vancomycin-intermediate resistance in Staphylococcus aureus. J. Glob. Antimicrob. Resist. 2014, 2, 213–224.
- Centers for Disease Control and Prevention. Staphylococcus aureus Resistant to Vancomycin—United States, 2002. MMWR Morb. Mortal. Wkly Rep. 2002, 51, 565–567.
- Liu, C.; Chambers, H.F. Staphylococcus aureus with Heterogeneous Resistance to Vancomycin: Epidemiology, Clinical Significance, and Critical Assessment of Diagnostic Methods. Antimicrob. Agents Chemother. 2003, 47, 3040–3045.
- Cameron, D.R.; Lin, Y.H.; Trouillet-Assant, S.; Tafani, V.; Kostoulias, X.; Mouhtouris, E.; Skinner, N.; Visvanathan, K.; Baines, S.L.; Howden, B.; et al. Vancomycin-intermediate Staphylococcus aureus isolates are attenuated for virulence when compared with susceptible progenitors. Clin. Microbiol. Infect. 2017, 23, 767–773.
- Jin, Y.; Yu, X.; Zhang, S.T.; Kong, X.Y.; Chen, W.W.; Luo, Q.X.; Zheng, B.W.; Xiao, Y.H. Comparative Analysis of Virulence and Toxin Expression of Vancomycin-Intermediate and Vancomycin-Sensitive Staphylococcus aureus Strains. Front. Microbiol. 2020, 11, 596942.
- Ohlsen, K.; Koller, K.P.; Hacker, J. Analysis of expression of the alpha-toxin gene (hla) of Staphylococcus aureus by using a chromosomally encoded hla::lacZ gene fusion. Infect. Immun. 1997, 65, 3606–3614.


