Enterococcus faecalis, Enterococcus faecium and Staphylococcus aureus are both common commensals and major opportunistic human pathogens. TIn recent decades, these bacteria have acquired broad resistance to several major classes of antibiotics, including commonly employed glycopeptides. Exemplified by resistance to vancomycin, glycopeptide resistance is mediated through intrinsic gene mutations, and/or transferrable van resistance gene cassette-carrying mobile genetic elements.
- antibiotic
- vancomycin
- drug-resistance
- Enterococcus
- Staphylococcus aureus
1. Introduction
1.1. Enterococcus faecalis and Enterococcus faecium
| Antibiotic Class | Resistance Gene(s), Family or Operon | Protein(s) Produced | Mechanism of Action | Gene Location(s) | Enterococcal Mobile Genetic Elements (MGEs) | References | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aminoglycosides | aac | Acetyltransferase | Antibiotic modification and inactivation | Chromosome, plasmid, transposon | Chromosome, plasmid, transposonPlasmids (P): pIP800, pJH1, pR538-1, pYN134, Inc. 18 Transposons (T): Tn1546, Tn4001, Tn5281, Tn5382, Tn5385 |
[36 | P: pETBTY825, pSK41, pUR1902, pUR2941 T: IS1181, IS1182, Tn4001, Tn5404, Tn5405 Tn554][ |
[39]37]37[40][38][41][39][42][40][43][41][44][42][138][43][139][44][140][45][141][46][142][47][143][48][144][49][145][50][[51][52][53],38[,39146][147][148][39,40,41,42,43,4454,40][,4155][36,,42,43,44,45,46,47,48,49,50,51,52,53,54,55] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| , | 138 | , | 139 | , | 140 | ,141,142,143,144,145,146,147,148] | aad, ant |
Adenylyltransferase | Plasmid, transposon | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| aad, ant | Adenylyltransferase | aph | Phosphotransferase | Plasmid, transposon | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| aph | Phosphotransferase | efmM | Methyltransferase | Methylation of 16S rRNA nucleotide; reduction of antibiotic target affinity | Chromosome | - | [56] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [ | 153 | ] | [ | 154 | ] | [155][156]][58,59[,145157],149[158],150[,151159,152,153,154,155,156,157,158,159] | Bacitracin, cephalosporins | croRS system | Penicillin-binding protein 5 (PBP5) and others | Cellular signaling in response to cell wall stress; deletion increases cellular susceptibility to antibiotics. Also involved in overexpression of PBP5 | Chromosome | - | [57] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cephalosporins, methicillin |
mecA | Penicillin-binding protein 2a (PBP2a) | Reduced antibiotic affinity; enables cell wall cross-linking in the presence of β-lactams | Chromosome, pathogenicity island (PAI) | PAI: SCCmec | [149][160][161][162][163][164][149,160,161,162,163,164] | β-lactams | blaZ | β-lactamase | Inactivation of β-lactam antibiotics through enzymatic hydrolysis | Chromosome, plasmid, transposon | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Chloramphenicol | cat | Chloramphenicol acetyltransferase | P: pBEM10 | T: Tn5385, Tn552 | [45][46][58][59][60][61][62][45,46,58, | Enzymatic acetylation of chloramphenicol; antibiotic inactivation59,60,61,62] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Plasmid | P: pC194, pC221, pUB112 | [ | 165 | ] | [ | 166][167][168][169][170][165,166,167,168,169,170] | Cephalosporin class β-lactams | pbp5 | Penicillin-binding protein | Reduced antibiotic affinity; enables cell wall cross-linking in the presence of β-lactams | Chromosome, plasmid, transposon | P: The possibility of plasmid-mediated pbp5 transfer has been mentioned. No pbp5-carrying plasmids have been described in E. faecalis or E. faecium, although it has been hypothesized.T: Conjugative transposon CTn5386 | [63][64][65][66][67][68][69][63,64,65,66,67,68,69] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IreK, ireP | IreK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Glycopeptides (e.g., vancomycin) | cls | Cardiolipin synthase | Involved in cell membrane synthesis; increased cls expression led to membrane modification that impaired antibiotic penetration and activity | Chromosome | - | [171][172][173][171,172,173] | —Ser/Thr kinaseireP—protein phosphatase | Part of a signaling transduction pathway that regulates cephalosporin resistance | Chromosome | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mprF | Bifunctional membrane enzyme involved in phospholipid synthesis and translocation | Increased positive charge of cell membrane and change of membrane fluidity that reduces antibiotic affinity; target modification | [ | - | 174][175][176][70] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [ | 174 | , | Chloramphenicol | cat | Chloramphenicol acetyltransferase |
Enzymatic acetylation of chloramphenicol; antibiotic inactivation | Plasmid | P: pRE25, pRUM, pIP501 | [71][72][73][74][71,72,73,74] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Glycopeptides (e.g., vancomycin) | liaFSR, liaXYZ | liaFSR is a regulatory system, with liaXYZ proteins being effector proteins | Modification of the cell membrane and envelope stress response. Modulates cell membrane localization or content, thus altering the antibiotic target site | Chromosome | - | [75][76][75,76] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cls | Cardiolipin synthase | Involved in cell membrane synthesis; increased cls expression led to membrane modification that impaired antibiotic penetration and activity | Chromosome | - | [77][78][79][77,78,79] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mprF | Bifunctional membrane enzyme involved in phospholipid synthesis and translocation | Increased positive charge of cell membrane and change of membrane fluidity that reduces antibiotic affinity; target modification | Homologs/paralogs described but not extensively studied—gene loci not reported | Unknown | P: Inc18-like, pLW1043, pSK41-like T: Tn1546[80][81][80,81] |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [ | 82 | ] | [ | 189 | ][190][191][192][193][82,189,190,191,192,193] | van operon (e.g., vanA) | van operon proteins—refer to | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fusidic acid | Section 2.3 | Reduction of antibiotic affinity through cell wall modification | Chromosome, plasmid, transposon | P: pHKK701, pHKK702, pHK703, pIP816, pIP964, pMG2200, pVEF1 T: Tn1546, Tn1547, Tn1549-like, Tn5482, Tn5506 |
[82][83][84][89] | fusA[ | Mutation to the EF-G ribosome complex90 | Antibiotic target modification; reduced antibiotic affinity][91][82,8385],84[86],85[87],86[88],87[,88,89,90,91] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Chromosome | - | [ | 194 | ] | Lincosamides Oxazolidinones Phenicols Pleuromutilins Streptogramin A |
cfr | rRNA methyltransferase | Methylation of A2503 bacterial 23S rRNA gene; reduced antibiotic affinity to methylated ribosomes | Chromosome, plasmid, transposon |
P: pEF-01 T: IS1216, Tn6218-like |
[92][ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| fusB | 93 | ] | FusB protein | [ | Prevention of antibiotic interaction with EF-G target site of bacterial ribosome | Chromosome, plasmid, transposon | P: pUB101 T: IS431/257 |
[194][195][196][194,195,196]94][92,93,94] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| optrA a | ABC-F protein | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| fusC | FusC protein | Active dislodgement of antibiotic from its ribosomal target site | Chromosome, plasmid, transposon | Chromosome, PAI | PAI: SCC476, SCCmecN1, pseudo SCCmec-SCC-SCCCRISPRP: Inc18, pE349
Table 2. Genetic basis of antibiotic resistance mechanisms in S. aureus .
a Confers resistance to oxazolidinones and phenicols only [99][136][99,136]. b Confers resistance to macrolides and streptogramins only [137]. c The fosB5 gene was not part of the IS257-like transposon but merely surrounded by two copies of it [231]. d A Nigerian study revealed a very low prevalence of plasmid-mediated qnr genes amongst clinical S. aureus isolates. No plasmid designations were provided from the study [236]. Quinolone resistance is caused primarily in Gram-negative bacteria through chromosomal mutations [252]. e tetS is carried by staphylococci [246] but has not been explicitly found in S. aureus in the literature.
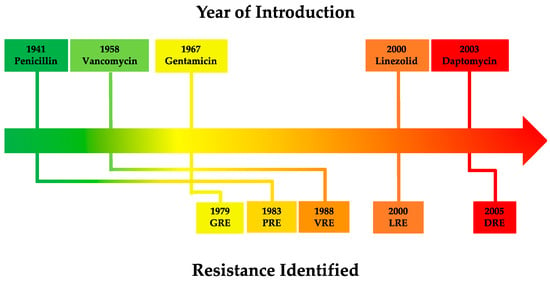
Vancomycin-resistant Enterococcus (VRE) is a frequent cause of clinical outbreaks worldwide [253][254][253,254]. An example of this is the VRE clonal sequence type 796 (ST796) which was first detected in 2011 in Australia, then quickly spread both nationwide and internationally to New Zealand [255] before also causing outbreaks in European hospitals beginning in December 2017 [256].
1.2. Staphylococcus aureusStaphylococcus aureus is a Gram-positive, facultative anaerobic bacterium [257][294]. Both a commensal as well as a significant pathogen of humans [258][295], S. aureus is prevalent in community, healthcare [259][296] and agricultural settings [260][261][297,298], asymptomatically colonising up to 30% of the human population [262][299].
As one of the most versatile and successful opportunistic human pathogens [259][263][296,300], S. aureus possesses a large variety of virulence factors [264][301] that enable host colonisation, tissue damage, immune evasion and progression of disease [264][265][301,302]. Consequently, S. aureus infections can be grouped into three general categories: (i) toxinoses such as scalded skin syndrome, food poisoning and toxic shock syndrome; (ii) benign and self-limiting conditions such as superficial skin and soft tissue infections; and (iii) systemic, life-threatening complications such as brain abscesses, meningitis, pneumonia, osteomyelitis, endocarditis, bacteremia, multi-organ failure and sepsis which carry high rates of morbidity and mortality [266][267][303,304].
S. aureus infections can be either HA or CA. The characteristics and virulence profiles of CA S. aureus typically differ to those of HA S. aureus [268][269][305,306]. HA infections of methicillin-resistant S. aureus (MRSA) were first reported in the 1960s [270][307], but rarely affected non-hospitalised healthy people and failed to spread efficiently within the community. This was generally attributed to the fitness cost imposed upon HA-MRSA through acquisition of antibiotic resistance elements [271][308], and is consistent with studies that reported HA-MRSA being generally more drug-resistant [268][272][305,309] and have reduced fitness and virulence [273][310] than CA-MRSA. Nevertheless, HA-MRSA clones remain a major cause of nosocomial infections globally [270][307].
The global emergence of CA-MRSA [274][275][276][277][278][279][311,312,313,314,315,316] began in the late 1980s [271][308], and was defined as a MRSA infection in the community whereby the infected persons exhibits no apparent nosocomial risk factors. This suggested that CA-MRSA evolved independently from lineages present in clinical settings. This hypothesis was further supported by the observation that CA-MRSA and HA-MRSA are epidemiologically, clinically, and microbiologically distinct [269][272][306,309]. Typically, CA-MRSA differ from HA-MRSA through the former exhibiting low-level susceptibility to non-β-lactam antibiotics, carriage of SCCmec types IV or V and production of Panton-Valentine leukocidins [271][308]. However, possible transmission between HA-MRSA to CA-MRSA may increase the overlap in similarities between the two MRSA sub-populations [269][306].
For HA-MRSA, ST239 was traditionally considered to be the dominant global hospital clone [280][317] and remains prevalent in Asia along with ST5 [281][282][318,319]. Elsewhere, the prevalence distribution of HA-MRSA clones will vary depending on geographical location: USA100 (North America) [283][284][285][286][320,321,322,323], CC5 (Latin America) [287][288][324,325], ST22 (United Kingdom), ST225 (central Europe) [282][319], ST22-IV [2B] (Australia) [289][326] and ST5 and ST239/241 (Africa) [290][327].
In the community, the dominant CA-MRSA clone also varies by geographical location: USA300—ST8-IV (North America), USA1100 and USA300-Latin American variant (South America), ST80-IV (Europe), ST93-IV (Australia), high heterogeneity in Asia (no dominant clone) and insufficient data for Africa [291][328]. Although traditionally considered a HA pathogen, the burden of CA-MRSA disease has been on the rise since its global emergence in the 1990s [292][329] and it began to appear within HA facilities in the 2000s [271][308]. Since then, many countries such as Australia [293][330], China [282][319], India [294][331], Kuwait [295][332], South Korea [296][297][333,334] Switzerland [298][335] United Arab Emirates [299][336], United Kingdom [300][337] and the United States [301][302][303][338,339,340] have reported the occurrences of persistence, dissemination, outbreaks and/or outright dominance of CA-MRSA clones within HA facilities which are attributed to the comparatively higher fitness of CA-MRSA through its carriage of smaller SCCmec variants and fewer, if any, other antibiotic resistance determinants [273][310].
The exact mechanisms driving the divergent evolution of MRSA clones, and reasons for the emergence and replacement or dominance of specific clones in different geographical locations remain unclear [270][274][304][305][306][307][307,311,341,342,343,344]. RWesearchers hypothesise that factors such as the host population demographics, migration, environmental climate, presence of other microorganism communities (e.g., other bacteria and bacteriophages that can facilitate horizontal gene transfer), spontaneous gene mutations and level of antibiotic use and stewardship are all likely to play contributing roles. With such changing diversity in MRSA clones, rapid and accurate clinical diagnosis, combined with a tailored treatment regimen according to the resistance profile of the clonal type will be essential for effective patient care [291][328].
S. aureus has demonstrated a remarkable ability to rapidly acquire and develop antibiotic resistance (Figure 2), often achieved through horizontal gene transfer of mobile genetic elements (MGEs) and chromosomal mutations [306][343]. As a result, an extensive arsenal of resistance mechanisms has emerged in S. aureus that enables resistance to major antibiotic classes typically employed to treat infection (Table 2).
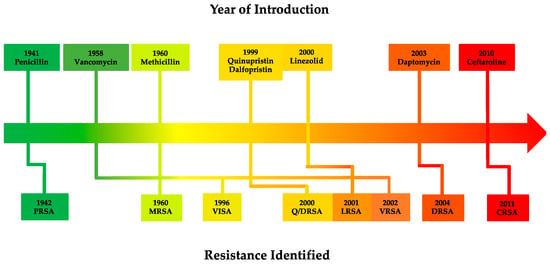 Figure 2. Timeline of antibiotic introduction (above) and subsequent resistance emergence in S. aureus (below) [29][30][33][34][308][309][29,30,33,34,345,346]. Abbreviations: PRSA—Penicillin-resistant S. aureus; MRSA—Methicillin-resistant S. aureus; VISA—Vancomycin-intermediate S. aureus; Q/DRSA—Quinupristin/dalfopristin-resistant S. aureus; LRSA—Linezolid-resistant S. aureus; VRSA—Vancomycin-resistant S. aureus; DRSA—Daptomycin-resistant S. aureus; CRSA—Ceftaroline-resistant S. aureus.
Like enterococci, the rapid emergence of resistance development in S. aureus has been attributed to the misuse and overuse of antibiotics in clinical and agricultural settings [9]. When combined with additional factors such as high rates of asymptomatic colonisation [262][310][299,347] and increased accessibility of international travel, S. aureus infections, particularly those caused by antibiotic resistant strains, have reached epidemic proportions in community and clinical settings worldwide [311][348]. Globally, S. aureus was responsible for more than 250,000 deaths associated with AMR in 2019 [312][268]. In the United States, S. aureus caused more than 119,000 bloodstream infections which led to nearly 20,000 deaths in 2017 [313][349]. As with VRE, antibiotic resistant S. aureus has also been designed as a “high priority” and “serious threat” pathogen by the WHO [314][274] and U.S. CDC [29] respectively.
2. Vancomycin2.1. Discovery and HistoryVancomycin is a tricyclic glycopeptide antibiotic first isolated in 1957 from the fungus Streptomyces orientalis. In vitro experiments showed that it had broad spectrum activity against Gram-positive bacteria, with no detected resistance in staphylococci following serial passages in media containing vancomycin. After showing promising efficacy and safety profiles in animal models, vancomycin (name derived from “vanquish”) entered human clinical trials [33][315][33,350]. During an initial clinical trial, vancomycin successfully treated 8 out of 9 patients with severe staphylococcal infection. Therapy failure occurred in one patient who was suffering from empyema, which prevented a therapeutic dose level of vancomycin from being administered [316][351]. In another human study, 5 out of 6 endocarditis patients who had already experienced antibiotic failure demonstrated resolution of disease indicators; the singular patient who experienced therapy failure had also presented with multiple conditions such as intractable heart failure and shock [317][352]. The culmination of positive data from these respective clinical trials subsequently resulted in the immediate approval of vancomycin by the U.S. Food and Drug Administration (FDA) in 1958. However, due to perceived nephrotoxicity [33][318][319][33,353,354], vancomycin was originally categorized as a last resort medication reserved for patients who were infected with bacteria that were resistant to frontline drugs or those patients with serious allergies to standard therapy [33]. Today, vancomycin is used as a first-line treatment for MRSA [320][321][322][355,356,357], and remains an important antibiotic used against serious Gram-positive bacterial infections [323][324][358,359].2.2. Mechanism of ActionVancomycin inhibits the cell wall synthesis of Gram-positive bacteria by binding to D-Ala-D-Ala dipeptide subunits of peptidoglycan monomers anchored to the sugar backbone of alternating N-acetylmuramic acid (MurNac) and N-acetylglucosamine (GlcNac) residues [325][326][360,361]. In susceptible bacteria, peptidoglycan monomers normally undergo transglycosylation and transpeptidation by the glycosyltransferase and transpeptidase activities of penicillin-binding proteins (PBPs), forming new peptidoglycan structures through pentaglycine cross-linkage [327][328][362,363]. Vancomycin, as a largely hydrophilic molecule, disrupts this process by forming hydrogen bonds to the D-Ala-D-Ala moiety through its aglycon subunit. As a result, this complex leads to a conformational change to the peptidoglycan which prevents subsequent transglycosylase and transpeptidase activity. Consequently, cell wall synthesis is inhibited as new peptidoglycan monomers are unable to be incorporated into the growing peptidoglycan skeleton, eventually leading to bacteriostasis in enterococci [315][350] or osmotic shock, cell lysis and death in S. aureus (Figure 3) [23][315][329][330][23,350,364,365].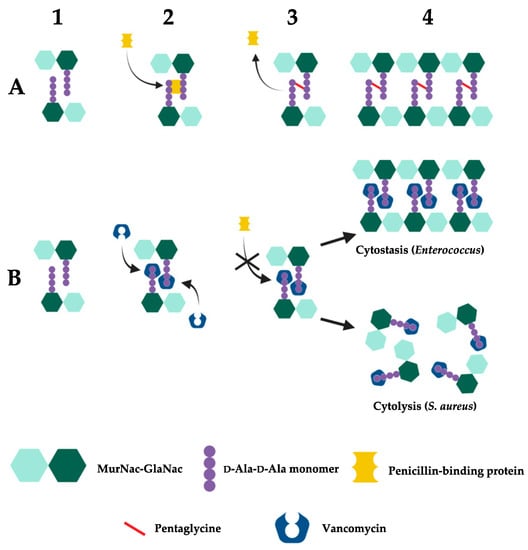 Figure 3. Mechanism of vancomycin activity. (A) Susceptible bacteria undergo normal cell wall synthesis through enzymatic (transglycosylase and transpeptidase) cross-linking activity in the absence of vancomycin. 1. Bacterial peptidoglycan with unlinked D-Ala-D-Ala monomers. 2. PBP recognises and binds to D-Ala-D-Ala monomers. 3. PBP facilitates the cross-linking of peptidoglycan D-Ala-D-Ala monomers through catalysis of pentaglycine bonds [23][315][325][326][327][328][329][23,350,360,361,362,363,364]. 4. Newly formed cell wall with complete cross-linking of D-Ala-D-Ala monomers. (B) Vancomycin inhibits peptidoglycan cross-linking in susceptible bacteria through its recognition and binding to D-Ala-D-Ala monomers. 1. Bacterial peptidoglycan with unlinked D-Ala-D-Ala monomers. 2. Vancomycin recognises and binds to D-Ala-D-Ala monomers. 3. Prevention of PBP-mediated catalysis of pentaglycine bonds due to vancomycin’s binding to D-Ala-D-Ala monomers. 4. Peptidoglycan cross-linking is inhibited, disrupting cell wall synthesis which leads to cytostasis (Enterococcus) or cell death (S. aureus) [23][315][325][326][327][328][329][23,350,360,361,362,363,364]. Created with BioRender.com.
2.3. Vancomycin Resistance in EnterococcusThe widespread use of vancomycin has predictably resulted in the rapid emergence and spread of vancomycin resistance amongst various Gram-positive bacteria [315][350]. In 1988, Uttley and colleagues published the first clinical outbreak of highly resistant VRE, with some isolates having minimum inhibitory concentrations (MICs) greater than 2000 µg/mL [332][367]. Today, the Clinical Laboratory Standards Institute (CLSI) classifies complete vancomycin resistance in Enterococcus with a MIC of ≥32 µg/mL, intermediate resistance as 8–16 µg/mL and susceptible as ≤4 µg/mL using broth microdilution testing [333][368]. This is consistent with the breakpoints set by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) which define the MIC of vancomycin susceptible enterococci to be ≤4 µg/mL and vancomycin-resistant enterococci to be>4 µg/mL [334][369]. Vancomycin resistance in enterococci is centered around the modification of the vancomycin target site i.e., modification of the D-Ala-D-Ala terminal amino acids of dipeptide monomer subunits into either D-Ala-D-Lac or D-Ala-D-Ser (Figure 4). These mutations confer high- and low-level vancomycin resistant phenotypes respectively. This is because the binding affinity of vancomycin for D-Ala-D-Lac is reduced 1000-fold due to loss of a single hydrogen bond [335][370] compared to its modestly (6-fold) reduced affinity for D-Ala-D-Ser due to steric hindrance by the D-Ser hydroxyl group [336][337][371,372]. As the mechanism of resistance (D-Ala-D-Lac or D-Ala-D-Ser) is determined by different van cassettes, the degree of vancomycin resistance in enterococci will be dependent upon which van operon they express [336][371]. The different van operons, their respective genes, proteins, and mechanisms of action responsible for these variable resistance levels are summarised in Table 3.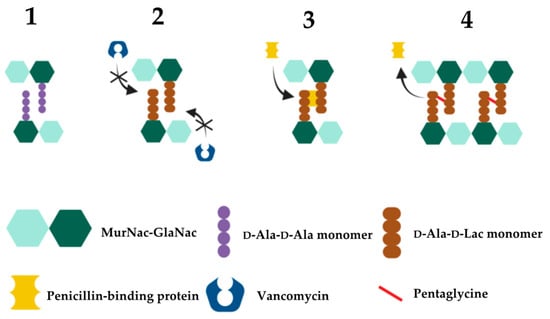 Figure 4. Mechanism of high-level vancomycin resistance. 1. Vancomycin-sensitive bacteria expressing D-Ala-D-Ala monomers. 2. Mutated vancomycin-resistant bacteria expressing D-Ala-D-Lac monomers that vancomycin poorly recognises. 3. With D-Ala-D-Lac not bound to vancomycin, PBP can subsequently bind to and catalyse the formation of pentaglycine bonds between MurNac-GlaNac monomers. 4. Bacterial peptidoglycan cross-linking and cell division continue uninhibited.
Table 3. The molecular basis of van-mediated vancomycin resistance in enterococci.
1vanA and vanC are the ligase genes of the vanA and vanC operons respectively. Ligase genes are named similarly for the other resistance cassettes e.g., vanB is the ligase gene designation for the vanB operon [336]. 2 The vanC operon is not found in E. faecium or E. faecalis [362]. 3 Not found in D-Ala-D-Lac based vancomycin resistance cassettes [336].  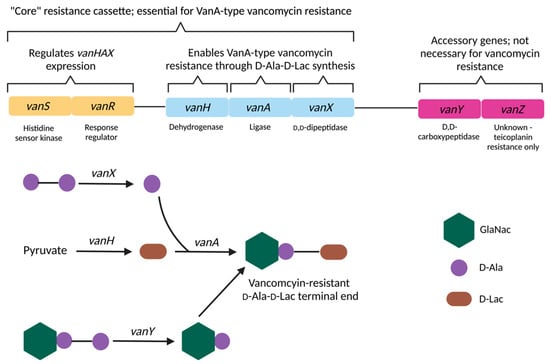 Figure 5. Molecular mechanisms of vanA-mediated vancomycin resistance. Expression of vanHAX genes are regulated by the two-component vanSR system. D-Ala-D-Ala components are hydrolysed by VanX, with VanY hydrolysing terminal D-Ala-D-Ala residues of existing peptidoglycan precursors not eliminated by VanX. Pyruvate is reduced to D-Lac by VanH, and VanA catalyses the esterification of D-Ala to D-Lac to form vancomycin-resistant peptidoglycan terminal ends. The role of VanZ in mediating vancomycin resistance is unknown and is implicated in teicoplanin resistance only [267][364][365][304,399,400]. Created with BioRender.com.
2.4. Vancomycin Resistance in S. aureusIn 1996, MRSA which were clinically refractory to vancomycin treatment was reported for the first time in Japan [308][345]. These strains, named Mu3 and Mu50, had MICs of 3 µg/mL and 8 µg/mL respectively [374][456] and were later termed hetero-vancomycin-intermediate S. aureus (hVISA) and vancomycin-intermediate S. aureus (VISA) respectively [375][457]. In 2002, complete VRSA was reported for the first time in the United States [376][458]. Today, CLSI defines S. aureus complete vancomycin resistance with a MIC of ≥16 µg/mL, intermediate resistance as 4–8 µg/mL and susceptible as ≤2 µg/mL with broth microdilution testing [333][368]. This is consistent with the breakpoints set by EUCAST which define the MIC of vancomycin susceptible S. aureus to be ≤2 µg/mL and vancomycin-resistant S. aureus to be >2µg/mL [334][369]. The conversion of vancomycin-susceptible S. aureus (VSSA) to VISA occurs through spontaneous mutations in genes such as walkR, rpoB, vraSR and mprF. Mutations in these genes are thought to confer the wide range of favorable phenotypic changes in VISA that allow for greater vancomycin resistance such as membrane charge modification, upregulation of cell wall biosynthesis genes, cell wall thickening, biofilm formation and modulation of key cellular processes such as immune evasion, virulence attenuation and reduced autolysis (Table 2) [174][175][176][177][178][179][180][181][182][183][184][185][186][187][188][375][174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,457]. hVISA is defined as the precursor to VISA, and in most cases emerges as a semi-resistant subpopulation of daughter cells from a previously susceptible but heterogenous VSSA population [375][457]. Following prolonged vancomycin exposure and selection pressure that favors their outgrowth, eventually a homogenous VISA population is achieved [377][459]. Interestingly, hVISA may also give rise to “slow” VISA (sVISA), a slower growing VISA subpopulation with similar phenotypes to extant “wild type” VISA but with longer doubling times, higher vancomycin MICs (≥6 µg/mL) and VISA phenotype instability (i.e., rapid reversion to hVISA in the absence of drug selection pressure) [375][457]. Despite the greater therapeutic difficulty in treating VISA infections, VISA appears to be less virulent than VSSA but with a greater ability to colonise and evade the host immune system [378][379][460,461]. Prior studies employing a mouse model of skin and soft tissue infection demonstrated that VISA had a much lower invasive capacity than VSSA; VISA also induced lower levels of innate immunity in persistent and chronic infections [379][461]. Proposed mechanisms for VISA’s virulence reduction include loss-of-activity mutations or dysfunction of the quorum-sensing, virulence regulator system agr [379][461]. Virulence factors under agr control include expression of the α-hemolysin encoded by hla [380][462], with mutant or dysfunctional agr VISA isolates found to produce up to 20-fold less α-toxin than VSSA and also less lethal in an in vivo murine bacteraemia model [378][460]. In addition, VISA had a comparatively higher capacity for biofilm formation than VISA, which are key contributors to S. aureus immune evasion and persistence. This was supported by the upregulation fnbB and sdrCDE genes which are associated with cell adhesion and immune evasion respectively [379][461].3. ConclusionE. faecalis, E. faecium and S. aureus are common human commensal organisms with potential to cause serious, life-threatening infections with exceptional intrinsic and acquired antibiotic resistance capabilities that have increased in prevalence globally in recent decades. This is enabled by the emergence and continued dissemination of mobile van resistance cassettes amongst staphylococci and enterococci through MGEs, which confer high-level vancomycin resistance through modification of the D-Ala-D-Ala peptidoglycan terminal ends in addition to endogenous, non-transferable mutations that give rise to intermediate-level resistance in S. aureus. Although other viable antibiotic combinations are still available for vancomycin resistant infections, novel antibiotics, in addition to alternative non-drug antimicrobial strategies will likely be needed to ensure treatment options remain for increasingly drug-resistant infections in the future. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||