Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Siva S. Panda | -- | 2787 | 2022-12-21 13:31:21 | | | |
| 2 | Conner Chen | -22 word(s) | 2765 | 2022-12-22 04:40:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Flint, A.L.; Hansen, D.W.; Brown, L.D.; Stewart, L.E.; Ortiz, E.; Panda, S.S. Anti-Breast Cancer Properties of Curcumin Analogs. Encyclopedia. Available online: https://encyclopedia.pub/entry/39047 (accessed on 25 June 2026).
Flint AL, Hansen DW, Brown LD, Stewart LE, Ortiz E, Panda SS. Anti-Breast Cancer Properties of Curcumin Analogs. Encyclopedia. Available at: https://encyclopedia.pub/entry/39047. Accessed June 25, 2026.
Flint, Abigail L., David W. Hansen, Lavauria D. Brown, Laura E. Stewart, Eduardo Ortiz, Siva S. Panda. "Anti-Breast Cancer Properties of Curcumin Analogs" Encyclopedia, https://encyclopedia.pub/entry/39047 (accessed June 25, 2026).
Flint, A.L., Hansen, D.W., Brown, L.D., Stewart, L.E., Ortiz, E., & Panda, S.S. (2022, December 21). Anti-Breast Cancer Properties of Curcumin Analogs. In Encyclopedia. https://encyclopedia.pub/entry/39047
Flint, Abigail L., et al. "Anti-Breast Cancer Properties of Curcumin Analogs." Encyclopedia. Web. 21 December, 2022.
Copy Citation
Breast cancer (BC), the most common malignancy in women, results from significant alterations in genetic and epigenetic mechanisms that alter multiple signaling pathways in growth and malignant progression, leading to limited long-term survival. Curcumin (a natural yellow pigment), the principal ingredient in the spice turmeric, is well-documented for its diverse pharmacological properties including anti-cancer activity. However, its clinical application has been limited because of its low solubility, stability, and bioavailability. To overcome the limitation of curcumin, several modified curcumin conjugates and curcumin analogs were developed and studied for their anti-cancer properties.
curcumin
curcumin mimic
conjugates
1. Introduction
Breast cancer is the most prevalent form of the disease found in women and is the main cause of cancer-related deaths in women globally. As with many other human cancers, breast cancer is caused by major modifications in genetic and epigenetic processes, as well as the targeting of many signaling pathways in the process of development and malignant progression toward an incurable and fatal disease. It has been established that a higher risk of breast cancer is related to both an earlier age at the onset of menarche as well as a later age at the onset of menopause. In the United States, roughly one in eight women at some point in their lives will be diagnosed with invasive breast cancer. This accounts for around thirteen percent of all women who are diagnosed with breast cancer. Furthermore, the Centers for Disease Control and Prevention (CDC) report breast cancer claims the lives of over 42,000 American women and 500 men each year [1][2]. As a result of this, research has been carried out to discover and develop drugs that are capable of effectively treating this particularly aggressive form of cancer. There is current difficulty in creating an effective treatment option for cancer, due to most available medications lacking the needed potency to provide full protection against the disease. The process of producing a new drug is time-consuming, challenging, and expensive. Additionally, it is intricate, and there is a high degree of unpredictability regarding the effectiveness of the drug after development. The difficulty in targeting cancer stem cells (CSCs), related to the drug-resistant properties of cancer stem cells rendering them immune to anti-cancer drugs, the lack of cancer epigenetic profiling, and the lack of specificity of existing epi-drugs are some of the most encountered challenges associated with cancer treatment [3].
Natural products have been used as a significant source of medications for many years. As of today, around half of all pharmaceuticals are still produced with the assistance of natural components. Several key commercialized medications in the field of cancer treatment have been derived from natural sources by structurally altering existing compounds or by synthesizing novel compounds like natural components. Research on modern anti-cancer drugs continues to place a significant emphasis on the search for improved cytotoxic agents. Due to the enormous structural diversity of natural compounds and the bioactivity potential of these compounds, several products that have been isolated from plants, marine flora, and microorganisms can serve as “lead” compounds. The improvement of their therapeutic potential through molecular modification can be carried out due to the large structural diversity that natural compounds possess [4].
Curcumin (Figure 1) is an example of a naturally occurring substance that has shown promise in treating breast cancer. Curcumin, a polyphenolic compound that can be found in turmeric (Curcumin longa), has been the topic of breast cancer research for over the past two decades due to its potential anti-inflammatory and anti-cancer effects. It has been revealed that curcumin suppresses the growth, spread, and metastasis of several malignancies. Its capacity to suppress oncogenic molecules such as protein kinases, cytokines, transcription factors, and growth factors plays a significant role in mediating its anti-cancer actions. In addition to this, curcumin obstructs the expansion and dissemination of cancer cells by obstructing their passage through the various phases of the cell cycle and/or by inducing apoptosis in cancer cells. However, according to pharmacokinetic studies, curcumin has poor systemic bioavailability because it is rapidly metabolized in the liver, where it undergoes glucuronidation and sulfation before being eliminated in the stool. The attempts undertaken up until this point to slow curcumin’s rapid metabolism have been ineffective in most cases. Due to curcumin’s restricted bioavailability and quick metabolism, researchers have previously explored and are currently exploring novel synthetic curcumin analogs that have lower toxicity, yet increased effectiveness [5][6].
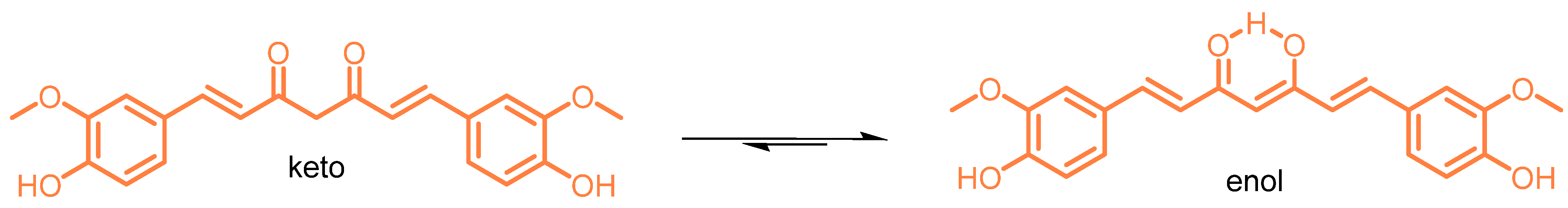
Figure 1. Keto-enol tautomeric forms of curcumin (CUR).
Curcumin scaffolds have been extensively explored and investigated including several analogs, conjugates, and mimics to reform their pharmacological potency and improve bioavailability [7][8][9][10][11]. The scaffold of curcumin has been broken down to identify the potential sites for structural modifications. The structure–activity relationship of the curcumin-based compounds indicates the key structural components/modifications responsible for a specific target (Figure 2). Curcumin itself has many pharmacological properties such as modulating signaling molecules, including cytokines, chemokines, transcription factors, adhesion molecules, microRNAs, tumor suppressor genes, etc. Structural modifications of curcumin have been advocated for improving its bioavailability, enhancing stability, and increasing potency. The modified curcumin could serve as the next generation of drug candidates for cancer therapy.
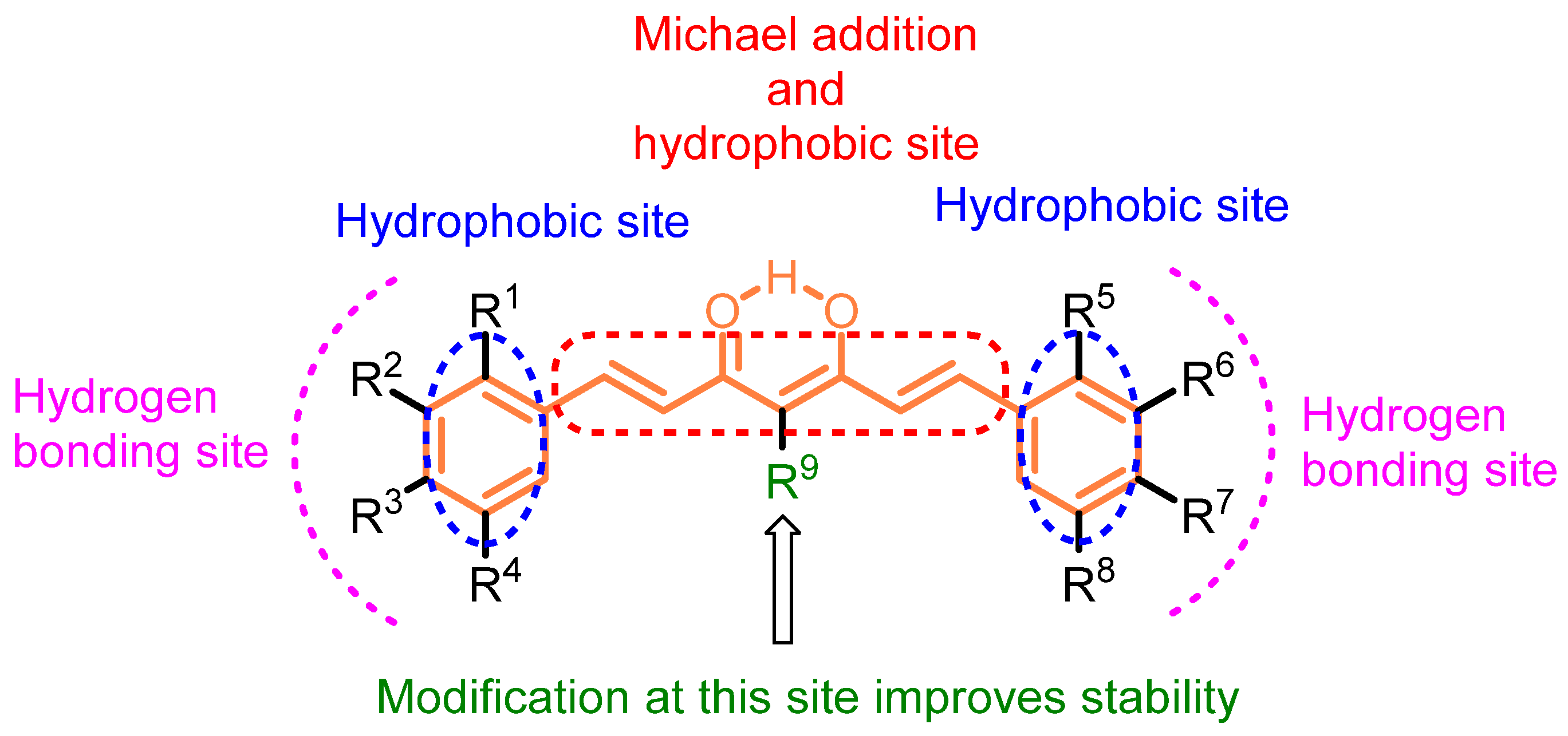
Figure 2. Structure of curcumin with indications of the important sites.
2. Anti-Breast Cancer Properties of Curcumin Analogs
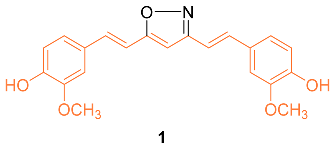
The isoxazole curcumin analog 1 was synthesized and the anti-cancer properties against the MCF-7 breast cancer cell line and its multidrug-resistant (MDR) version, MCF-7R, were compared with curcumin. After 72 h of treatment, the IC50 of curcumin was calculated from four separate experiments to be 29.3 ± 1.7 μM in MCF-7 and 26.2 ± 1.6 μM in MCF-7R, indicating that the cytotoxic activity of curcumin in the MDR breast cancer cell line is at least equivalent to, and perhaps slightly stronger than, its parental variant. In both the parental and MDR cell line, derivative 1 was more effective than curcumin with an IC50 of 13.1 ± 1.6 μM in MCF-7 and 12.0 ± 2.0 μM in MCF-7R. An MDR form of HL-60 leukemia also showed comparable outcomes. RT-PCR analyses in MCF-7 and MCF-7R cell lines revealed that curcumin and 1 caused early changes in the quantities of important gene transcripts, which were, nevertheless, primarily varied between the two cell lines. Overall, these results show that the expression of P-gp or the absence of ER in breast cancer cells does not impede the anti-cancer activities of either curcumin or 1. Remarkably, the agents seemed to adjust their molecular actions in response to the different patterns of gene expression found in the MDR and the parental MCF-7 [12].
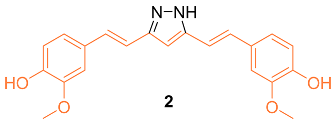
According to Wang et al., research was conducted on hydrazinocurcumin 2 (HC) to investigate its effectiveness against breast cancer cells, specifically in the cell lines MDA-MB-231 and MCF-7. After 72 h of treatment, dose-dependent suppression of tumor cell survival and proliferation was seen for the MDA-MB-231 and MCF-7 cell lines. The IC50 values for 2 were 3.37 μM and 2.57 μM, respectively, which were both significantly lower than those for curcumin (26.9 μM and 21.22 μM). Compared to curcumin, the results demonstrated that 2 was significantly more effective in suppressing cell viability in both cell lines tested. Apoptosis was induced in MDA-MB-231 and MCF-7 cells using FCM, and the influence of 2 and curcumin on this process was analyzed. At 10 µM, 2 significantly induced cells apoptosis (14% in MDA-MB-231 cells and 26% in MCF-7 cells), whereas at the same concentration, curcumin only induced 9% and 20% cell apoptosis in MDA-MB-231 and MCF-7 cells, respectively. The results showed that 2 caused an increase in the apoptotic rate of cells in a dose-dependent manner after a treatment period of 48 h. In addition, the Western blot analysis demonstrated that 2 was much more effective than curcumin in suppressing the production of STAT3 protein in MDA-MB-231 and MCF-7 cells at the same concentration (10–20 μM). The data showed that 2 was more effective than curcumin at suppressing cell proliferation, losing colony formation, depressing cell migration and invasion, and inducing cell death via inhibiting STAT3 phosphorylation and downregulating an array of STAT3 downstream targets [13].
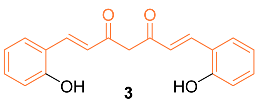
Mohankumar et al. studied the apoptotic mechanism of 3, an ortho-hydroxy substituted analog of curcumin using an in vitro and in silico approach. In the study, it was found that 3 exhibited a greater potency in the modulation of selective apoptotic markers and inhibited MCF-7 at a dose level of 30 µM (equivalent dosage level to curcumin), and significantly regulated PI3k/Akt, both intrinsic and extrinsic apoptotic pathways, by inhibiting Bcl-2 and inducing p53, Bax, cytochrome c, Apaf-1, FasL, caspases-8, 9, 3, and PARP cleavage. mRNA expression studies for Bcl-2/Bax indicated increased efficiency with 3 compared to curcumin, while an in silico molecular docking study utilizing PI3K revealed that the docking of 8 was more potent than curcumin. Cells treated with 3 effectively induced apoptosis through ROS intermediates, as measured by 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). Results showed 3 induced apoptosis more effectively than curcumin, and this activity can be attributed to the presence of the hydroxyl group in the ortho position in the structure [14]. In addition, Western blotting indicated that compound 3 significantly downregulated the expression levels of NF-κB, p65, and c-Rel. In addition, src levels were significantly reduced in comparison to cells treated with curcumin. In silico docking studies were performed with the derivative and curcumin with NF-κB (PDB ID: 1NFK). The results indicated that the derivative displayed a stronger interaction with NF-κB compared to curcumin, with a Lidblock score of 109.814 while curcumin’s was 95.696 [15].
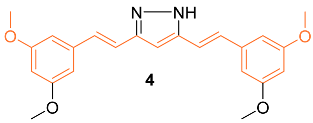
Lien et al. synthesized over 30 curcumin derivatives and published findings that a novel curcumin derivative (4) inhibits cell proliferation and drug resistance of HER2-overexpressing cancer cells. The mimic was tested in vitro on both the MCF-7 and MDA-MB-435 cell lines transfected with pSV2-erbB2. Results indicated that the derivative preferentially suppresses the growth of HER2-overexpressing cancer cells. Studies were also carried out to investigate if the derivative would sensitize HER2-overexpressing cancer cells to clinical drugs and it was found that overexpressing cells showed greater cytotoxic activity when the derivative was administered in combination with doxorubicin (DOX), etoposide, or taxol [16].
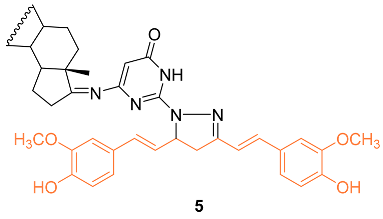
To understand the molecular hybridization impact and the integration of two drugs with different modes of action, affecting the same target, a variety of heterocyclic steroids and curcumin moieties were considered for the synthesis of hybrid conjugates and to determine their anti-cancer activity. The authors synthesized the hetero-steroid compounds and conducted in vitro studies of the cytotoxic effects against the MCF-7 breast cancer cell line. Of all compounds, 5 had the best cytotoxic activity against the MCF-7 cell line with an IC50 value of 18 μM. This compound is also promising as an anti-cancer compound having pro-apoptotic effects resulting in desired cell growth inhibition [17].
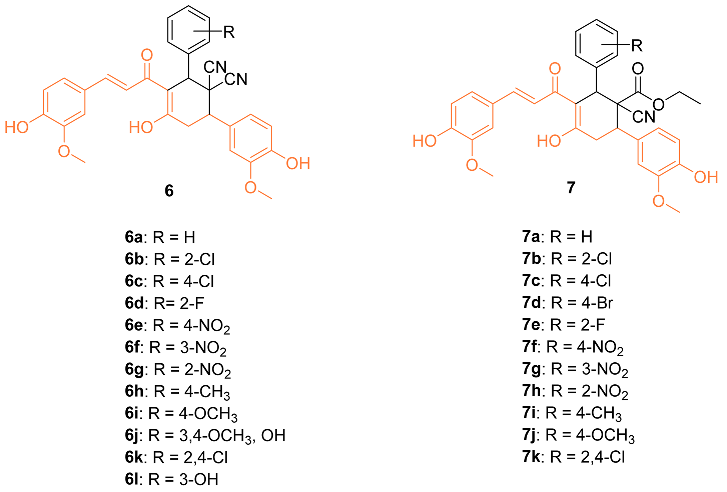
Bhuvaneswari et al. reported the biological evaluation and molecular docking of novel curcumin derivatives 6a–l and 7a–k. Firstly, in vitro cytotoxicity was tested against the MCF-7 breast cancer cell line. The IC50 for 6j and 7i were 15 µM and 10 µM, respectively. When the compounds were tested against normal HBL-100 cells, the cells were resistant to the compounds up to 50 µM doses, showing the compounds are selective and dose-dependent. Molecular docking studies were also conducted in PatchDock and suggested that these two compounds could be the starting point for designing new potent Bcl-2 anti-apoptotic protein inhibitors, with 7 having a geometrical score of 6028 and 6 with a score of 5962 compared to 4190 for curcumin (PDB: 1GJH) [18].
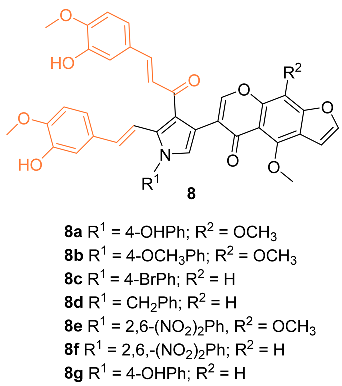
Nagwa et al. synthesized a set of curcumin derivatives 8a–g and then experimented to determine the efficacy of the derivatives against breast cancer. Preliminary tests were conducted with normal MCF-10A cells and it was found that all derivatives had little cytotoxicity, with more than 85% cell viability. An MTT assay was performed with the derivatives against an MCF-7 breast cancer cell line. Compounds 8a and 8c were the most potent against the breast cancer cells, with an IC50 of 20 and 22 μg/mL, respectively. Pharmacokinetic (ADME) studies confirm that compounds 8a and 8c have good intestinal absorption and are non-carcinogenic [19].
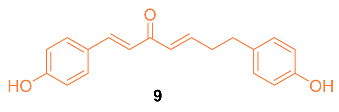
Hong et al. reported the synthesis of and anti-cancer studies on the novel curcumin mimic (1E,4E)-1,7-bis(4-hydroxyphenyl) (hepta-1,4-dien-3-one) 9 isolated from mistletoe. It was first tested for in vitro cytotoxicity in which it showed activity in the micromolar range. Additionally, 9 showed a higher potency than cis-platinum against four human breast cancer cell lines (SKBR3, MDA-MB231, MCF-7, and MDA-MB453). The IC50 values for the breast cancer cell lines were significantly lower with 9 compared to cis-platinum. The cytotoxicity of 9 with normal cells was investigated with LO2 human liver cells, GES-1 human gastric epithelial cells, and BEAS-2B human lung epithelial cells. The results indicated that 9 had a little inhibitory effect on normal cells, with each group having a less than 5% inhibition rate, which is much lower than the rate on cancer cells at the same concentration, indicating 9 has a selectivity for the toxic effects of cancer cells rather than normal cells. In addition, in vivo data on the MCF-7 breast cancer model in mice suggest that 9 is more effective than cisplatin. The groups administered 9 had a stable weight for up to 9 days, while a clear weight loss was observed in the positive control group [20].
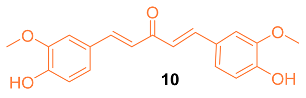
Shen et al. tested the efficacy of a curcumin analog 10 in breast cancer cells. The breast cancer cell lines MCF-7 and MDA-MB-231 were used to study the cell viability, cell migration, cell cycle, and apoptosis of this analog. It was shown that when the concentration of 10 was increased, there was a decrease in cell viability. In addition, 10 had an IC50 of 8.84 μM compared to curcumin with an IC50 of 16.85 μM against MCF-7 breast cancer cells. It was shown that 10 is a compound that activates the mitochondrial apoptosis pathway in breast cancer cells [21].
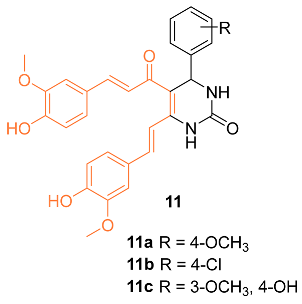
Sharma et al. synthesized 3,4-Dihydropyrimidin-2(1H)-one/thione curcumin analogs and, among them, compounds 11a–c were submitted to the National Cancer Institute (NCI) to investigate activity against various cell lines, including the breast cancer cell lines MDA-MB-231 and HS 578T. At a concentration of 100 µM, compounds 11a–c all displayed moderate activity, with compound 11a being the most active. This is supported by a growth percent value (GP) of 55.45 for compound 11a on MDA-MB-231 cells and a GP of 73.39 on HS 578T cells, while activities with a GP of 73.63 and 67.70 on MDA-MB-231 cells were found for compounds 11b and 11c, respectively. The authors believe compounds 11a–c should be further studied to increase the moderate anti-cancer activity [22].
Zhang et al. reported on the synthesis and anti-cancer activity of ten curcumin mimics 12a–j. Compound 12b exhibited the best anti-cancer activity, with an IC50 value of 4.99 µM against MDA-MB-231 breast cancer cells compared to the 6.18 µM of cisplatin. In vivo data were obtained and were promising. However, in vivo testing was only carried out on H22 hepatic cells. Further testing is needed to evaluate if compound 12b is a promising anti-breast cancer drug [23].
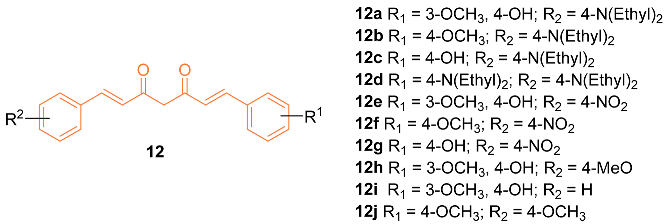
Considering the importance of pyrazole moiety, Ahsan et al. synthesized various curcumin analogs 13–15 containing a pyrazole or pyrimidine ring to target the epidermal growth factor receptor (EGFR) tyrosine kinase. Fourteen curcumin analogs with pyrazole or pyrimidine moieties were synthesized, with ten being evaluated amongst 60 different cell lines to observe anti-cancer effects. The activity was observed from various compounds, however, 13–15 displayed anti-cancer activity against various cell lines including MDA-MB-468. Compound 13 showed a cell promotion of −30.34%, compound 14 showed −31.86%, and compound 15 showed −35.04%. Ahsan et al. claim their curcumin analogs are promising and can be a therapeutic intervention in cancer treatment [24].
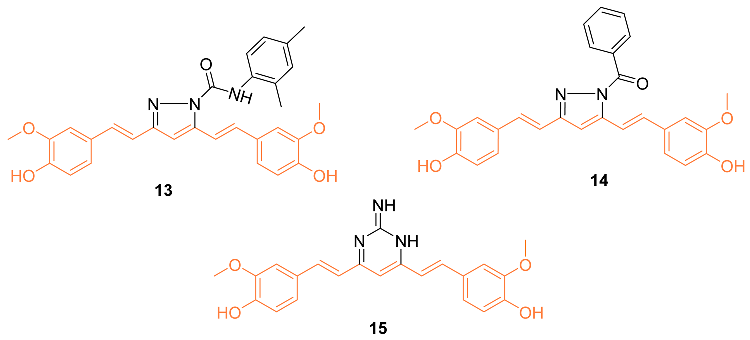
A set of twenty-four different analogs of curcumin containing pentadienone moiety were synthesized and examined for their anti-cancer properties against breast cancer cells (MCF-7 and MDA-MB-231). A dose-dependent suppression of tumor cell survival and proliferation was observed after 72 h of treatment with compounds 16–18. The IC50 values for compound 16 were 2.7 ± 0.5 μM and 1.5 ± 0.1 μM for the cell lines MCF-7 and MDA-MB-231, respectively, which were 5-8 times lower than those for curcumin (21.5 ± 4.7 μM and 25.6 ± 4.8 μM). Furthermore, the IC50 values of compounds 17 (0.4 ± 0.1 μM and 0.6 ± 0.1 μM) and 18 (2.4 ± 1.0 μM and 2.4 ± 0.4 μM) were favorable for the MCF-7 and MDA-MB-231 cell lines, respectively. The non-malignant mammary epithelial cell line (MCF-10) demonstrated no toxicity from any of the three compounds. In comparison to curcumin, which did not exhibit any selectivity against cancer cell lines, it was discovered that compounds 16–18 displayed a selectivity ratio of at least fivefold or greater. Compound 17, having IC50 values in the sub-micromolar range and a selectivity ratio greater than 25, was found to be the most potent analog. All three compounds, however, show promise as possible anti-tumor drug candidates for breast cancer [25].
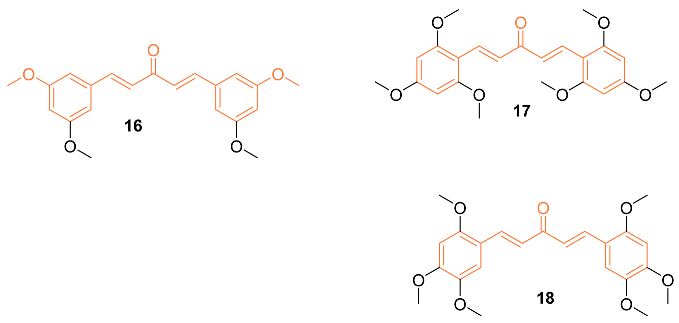
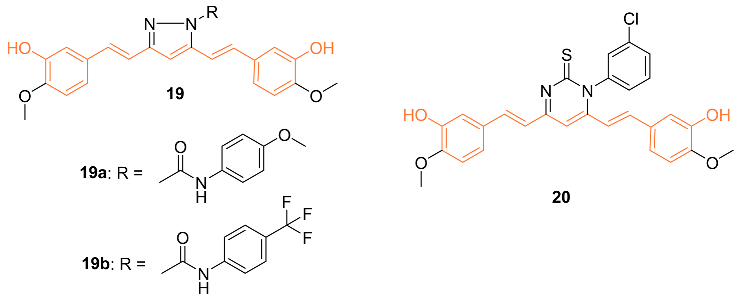
Ali et al. synthesized curcumin analogs 19a, 19b, and 20 and tested them against several breast cancer lines to determine their anti-cancer effects. The compounds were docked against the epidermal growth factor receptor, which allowed for the determination of binding efficiency. All derivatives showed moderate inhibition of epidermal growth factor receptors. Compounds 19b and 20 showed the most anti-cancer activity against BT-549 with GI50 values of 2.98 μM for 2 and 1.51 μM for 3 [26].
References
- Breast Cancer Facts and Statistics. Available online: https://www.breastcancer.org/facts-statistics (accessed on 11 October 2022).
- Basic Information About Breast Cancer. Available online: https://www.cdc.gov/cancer/breast/basic_info/index.htm#:~:text=About%2042%2C000%20women%20and%20500,breast%20cancer%20than%20White%20women (accessed on 11 October 2022).
- Chakraborty, S.; Rahman, T. The Difficulties in Cancer Treatment. Ecancermedicalscience 2012, 6, 1–5.
- Gordaliza, M. Natural Products as leads to anticancer drugs. Clin. Trans. Oncol. 2007, 9, 767–776.
- Panda, S.S.; Girgis, A.S.; Thomas, S.J.; Capito, J.E.; George, R.F.; Salman, A.; El-Manawaty, M.A.; Samir, A. Synthesis, pharmacological profile and 2D-QSAR studies of curcuminamino acid conjugates as potential drug candidates. Eur. J. Med. Chem. 2020, 196, 112293.
- Vyas, A.; Dandawate, P.; Padhye, S.; Ahmad, A.; Sarkar, F. Perspectives on New Synthetic Curcumin Analogs and their Potential Anticancer Properties. Curr. Pharm. Des. 2013, 19, 2047–2069.
- Yin, Y.; Tan, Y.; Wei, X.; Li, X.; Chen, H.; Yang, Z.; Tang, G.; Yao, X.; Mi, P.; Zheng, X. Recent Advances of Curcumin Derivatives in Breast Cancer. Chem. Biodivers. 2022, 19, e202200485.
- Awasthi, M.; Singh, S.; Pandey, V.P.; Dwivedi, U. Curcumin: Structure-Activity Relationship Towards its Role as a Versatile Multi-Targeted Therapeutics. Mini Rev. Org. Chem. 2017, 14, 311–332.
- Moreira, J.; Saraiva, L.; Pinto, M.M.; Cidade, H. Diarylpentanoids with antitumor activity: A critical review of structure-activity relationship studies. Eur. J. Med. Chem. 2020, 192, 112177.
- Gupta, A.P.; Khan, S.; Manzoor, M.M.; Yadav, A.K.; Sharma, G.; Anand, R.; Gupta, S. Chapter 10—Anticancer Curcumin: Natural Analogues and Structure-Activity Relationship. Stud. Nat. Prod. 2017, 54, 355–401.
- Rodrigues, F.C.; Kumar, N.A.; Thakur, G. Developments in the anticancer activity of structurally modified curcumin: An up-to-date review. Eur. J. Med. Chem. 2019, 177, 76–104.
- Poma, P.; Notarbartolo, M.; Labbozzetta, M.; Maurici, A.; Carina, V.; Alaimo, A.; Rizzi, M.; Simoni, D.; D’Alessandro, N. The antitumor activities of curcumin and of its isoxazole analogue are not affected by multiple gene expression changes in an MDR model of the MCF-7 breast cancer cell line: Analysis of the possible molecular basis. Int. J. Mol. Med. 2007, 20, 329–335.
- Wang, X.; Zhang, Y.; Zhang, X.; Tian, W.; Feng, W.; Chen, T. The curcumin analogue hydrazinocurcumin exhibits potent suppressive activity on carcinogenicity of breast cancer cells via STAT3 inhibition. Int. J. Oncol. 2012, 40, 1189–1195.
- Mohankumar, K.; Pajaniradje, S.; Sridharan, S.; Singh, V.K.; Ronsard, L.; Banerjea, A.C.; Benson, C.S.; Coumar, M.S.; Rukkumani, R. Mechanism of apoptotic induction in human breast cancer cell, MCF-7, by an analog of curcumin in comparison with curcumin—An in vitro and in silico approach. Chem. Biol. Interact. 2014, 210, 51–63.
- Mohankumar, K.; Sridharan, S.; Pajaniradje, S.; Singh, V.K.; Ronsard, L.; Banerjea, A.C.; Somasundaram, D.B.; Coumar, M.S.; Periyasamy, L.; Rajagopalan, R. BDMC-A, an analog of curcumin, inhibits markers of invasion, angiogenesis, and metastasis in breast cancer cells via NF-kB pathway—A comparative study with curcumin. Biomed. Pharmacother. 2015, 74, 178–186.
- Lien, J.C.; Hung, C.M.; Lin, Y.J.; Lin, H.C.; Ko, T.C.; Tseng, L.C.; Kuo, S.C.; Ho, C.T.; Lee, J.C.; Way, T.D. Pculin02H, a curcumin derivative, inhibits proliferation and clinical drug resistance of HER2-overexpressing cancer cells. Chem. Biol. Interact. 2015, 235, 17–26.
- Elmegeed, G.A.; Yahya, S.M.M.; Abd-Elhalim, M.M.; Mohamed, M.S.; Mohareb, R.M.; Elsayed, G.H. Evaluation of heterocyclic steroids and curcumin derivatives as anti-breast cancer agents: Studying the effect on apoptosis in MCF-7 breast cancer cells. Steroids 2016, 115, 80–89.
- Bhuvaneswari, K.; Sivaguru, P.; Lalitha, A. Synthesis, Biological Evaluation and Molecular Docking of Novel Curcumin Derivatives as Bcl-2 Inhibitors Targeting Human Breast Cancer MCF-7 Cells. ChemistrySelect 2017, 2, 11552–11560.
- Nagwa, M.F.; Sarhan, A.E.; Elhefny, E.A.; Nasef, A.M.; Aly, M.S. Synthesis, Cytotoxicity Evaluation, and Molecular Docking Studies of Novel Pyrrole Derivatives of Khellin and Visnagin via One-Pot Condensation Reaction with Curcumin. Russ. J. Bioorg. Chem. 2020, 46, 1117–1127.
- Hong, J.; Meng, L.; Yu, P.; Zhou, C.; Zhang, Z.; Yu, Z.; Qin, F.; Zhao, Y. Novel drug isolated from mistletoe (1E,4E)-1,7-bis(4-hydroxyphenyl)hepta-1,4-dien-3-one for potential treatment of various cancers: Synthesis, pharmacokinetics and pharmacodynamics. RSC Adv. 2020, 10, 27794–27804.
- Shen, H.; Shen, J.; Pan, H.; Xu, L.; Sheng, H.; Liu, B.; Yao, M. Curcumin analog B14 has high bioavailability and enhances the effect of anti-breast cancer cells in vitro and in vivo. Cancer Sci. 2021, 112, 815–827.
- Sharma, R.; Jadav, S.S.; Yasmin, S.; Bhatia, S.; Khalilullah, H.; Ahsan, M.J. Simple, efficient, and improved synthesis of Biginelli-type compounds of curcumin as anticancer agents. Med. Chem. Res. 2015, 24, 636–644.
- Zhang, L.; Zong, H.; Lu, H.; Gong, J.; Ma, F. Discovery of novel anti-tumor curcumin analogues from the optimization of curcumin scaffold. Med. Chem. Res. 2017, 26, 2468–2476.
- Ahsan, M.J.; Khalilullah, H.; Yasmin, S.; Jadav, S.S.; Govindasamy, J. Synthesis, Characterisation, and In Vitro Anticancer Activity of Curcumin Analogues Bearing Pyrazole/Pyrimidine Ring Targeting EGFR Tyrosine Kinase. BioMed Res. Int. 2013, 2013, 239354.
- Fuchs, J.R.; Pandit, B.; Bhasin, D.; Etter, J.P.; Regan, N.; Abdelhamid, D.; Li, C.; Lin, J.; Li, P. Structure-activity relationship studies of curcumin analogues. Bioorg. Med. Chem. Lett. 2009, 19, 2065–2069.
- Ali, A.; Ali, A.; Tahir, A.; Bakht, A.M.; Salahuddin; Ahsan, M.J. Molecular Engineering of Curcumin, an Active Constituent of Curcuma longa L. (Turmeric) of the Family Zingiberaceae with Improved Antiproliferative Activity. Plants 2021, 10, 1559.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
787
Revisions:
2 times
(View History)
Update Date:
22 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No