Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Olga A. Korczeniewska | -- | 3909 | 2022-12-14 19:29:20 | | | |
| 2 | Sirius Huang | + 4 word(s) | 3913 | 2022-12-15 03:51:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Korczeniewska, O.A.; Kohli, D.; Benoliel, R.; Baddireddy, S.M.; Eliav, E. Peripheral Events in the Pathophysiology of PTNP. Encyclopedia. Available online: https://encyclopedia.pub/entry/38785 (accessed on 06 June 2026).
Korczeniewska OA, Kohli D, Benoliel R, Baddireddy SM, Eliav E. Peripheral Events in the Pathophysiology of PTNP. Encyclopedia. Available at: https://encyclopedia.pub/entry/38785. Accessed June 06, 2026.
Korczeniewska, Olga A., Divya Kohli, Rafael Benoliel, Sita Mahalakshmi Baddireddy, Eli Eliav. "Peripheral Events in the Pathophysiology of PTNP" Encyclopedia, https://encyclopedia.pub/entry/38785 (accessed June 06, 2026).
Korczeniewska, O.A., Kohli, D., Benoliel, R., Baddireddy, S.M., & Eliav, E. (2022, December 14). Peripheral Events in the Pathophysiology of PTNP. In Encyclopedia. https://encyclopedia.pub/entry/38785
Korczeniewska, Olga A., et al. "Peripheral Events in the Pathophysiology of PTNP." Encyclopedia. Web. 14 December, 2022.
Copy Citation
The ICOP (International Classification of Orofacial Pain), 1ST edition describes post-traumatic trigeminal neuropathic pain (PTNP) as “a unilateral or bilateral facial or oral pain following and caused by trauma to the trigeminal nerve(s), with other symptoms and/or clinical signs of trigeminal nerve dysfunction, and persisting or recurring for more than 3 months”. A cascade of events in the peripheral and central nervous system function is involved in the pathophysiology of pain following nerve injuries. Central and peripheral processes occur in tandem and may often be co-dependent. Due to the complexity of central mechanisms, only peripheral events contributing to the pathophysiology have been reviewed herein.
neuropathic pain
trigeminal nerve injury
pathophysiology
trauma
1. Introduction
A cascade of events in the nervous system function is involved in the pathophysiology of pain following nerve injuries. The current understanding of these events is primarily based on the findings from animal research that focused predominantly on injuries to the spinal nerves with limited number of studies investigating trigeminal nerve injuries induced neuropathies. The events following nerve injury are time-dependent, progressing from the peripheral to the central nervous system and include changes in the functional, biochemical and physical characteristics of neurons and glial cells [1][2][3]. As mentioned earlier, this text will focus mainly on peripheral events contributing to the pathophysiology of post-traumatic trigeminal neuropathic pain (PTNP), however, both peripheral and central processes play a role in the neuropathy development and occur in tandem.
2. The Role of Receptors and Inflammatory Soup
Nerve injury is accompanied by a release of mediators such as bradykinin, prostaglandin E2, serotonin (5HT), ATP and H+ that trigger their corresponding receptors that activate intracellular processes via protein kinase A and C (Figure 1). The activated protein kinases phosphorylate transducer channels, leading to a reduction of thresholds and enhanced excitability of membrane on the peripheral terminals [4][5][6]. The phosphorylation of voltage gated sodium channels (Nav1.8 and Nav1.9) produces enhanced currents and hyperexcitability characterized by reduction of activation threshold and an increased firing rate [7][8][9][10]. Injury to the trigeminal nerve has also been shown to cause a reduction in voltage gated K+ channels (transient and sustained: IA and IK) current in the trigeminal ganglion neurons. This causes an increase in the spike frequency and duration and decrease in activation threshold [11][12]. Nerve injury secondary to trauma or disease initiates inflammatory processes, alters electrical activity of neurons, and allows for cross talk between neurons resulting in phenomenon called peripheral sensitization. The substances released during tissue injury initiate a cascade of events that may lead to nociceptors’ activation sometimes even in the absence of a noxious stimulus.
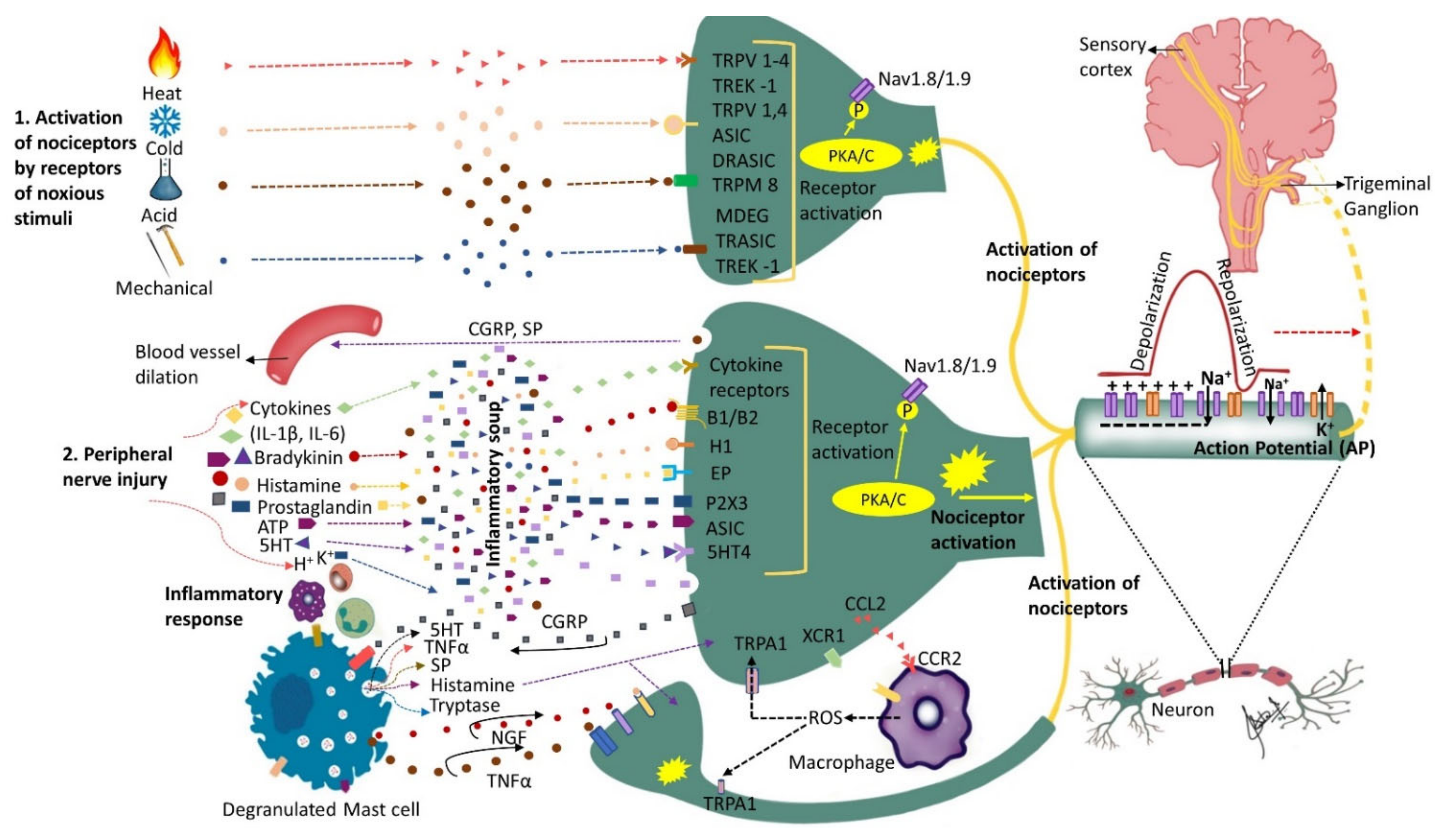
Figure 1. Nociceptors activation and peripheral sensitization: Nociceptors are a specialized class of primary afferents that respond to noxious stimuli. Some nociceptors are activated by noxious stimuli (heat, cold, mechanical or chemical) whereas others respond to substances that are released following tissue injury (cytokines, bradykinin, histamine, endothelin-1, ATP, prostaglandins, etc.). The K+ ions released by neighboring injured cells directly depolarize nociceptor membranes. Trunsduction of noxious stimuli (thermal, mechanical or chemical) into an electric potential or binding of the inflammatory mediators secreted by the injured cells to their corresponding receptors sets off a chain of events leading to nociceptor sensitization and peripheral sensitization. Peripheral sensitization results in lower activation threshold and higher firing rate. Upon activation, nociceptors release substance P and calcitonin-gene-related peptide (CGRP) that initiate inflammatory response including blood vessel dilation and mast cell degranulation (activation). Activated mast cells release histamine, serotonin, nerve growth factor (NGF), substance P (SP) and tumor necrosis factor alpha (TNF-α) that bind to their corresponding receptors on the nociceptor, further sensitizing the nociceptor. Activated nociceptors generate an electric potential. Nerve injury induces further molecular changes, including an increase in sodium (Na+) channels and a decrease in potassium (K+) channels further sensitizing the nociceptor and triggering ectopic activity, as well as alterations in the trigeminal ganglion. Additionally, injured nerves release chemokine CCL2 that attracts macrophages to the site of injury further promoting neuropathic pain development. This figure illustrates only the ascending pathway and not additional pathways that may contribute to the input from the peripheral to the central nervous system.
3. The Role of Chemokines
Chemokine are key players in pain modulation in the peripheral and central nervous systems [13]. Their role in trigeminal pain and their pivotal role in PTNP development has been reviewed recently elsewhere [14]. CCL2 is a proinflammatory chemokine that recruits monocytes, memory T cells and dendritic cells to the site of inflammation [15] that is also known to be a glial cells mediator [16]. It is released from damaged nerves [17] as a direct consequence of the early afferent activity which is critical for the development of neuropathic pain [18]. Released CCL2 promotes increase in Nav1.8 sodium channel activity in primary sensory neurons through a Gβγ-dependent mechanism [19]. Additionally, released CCL2 attracts macrophages to the site of injury [20] by binding to its receptor (CCR2) on macrophages. Macrophages have been shown to promote neuropathic pain development by neuro-immune interactions and their absence was shown to prevent the development of neuropathic pain [21][22]. Other chemokines such as, CXCL13 and its receptor CXCR5 have been implicated in trigeminal neuropathic pain induced by nerve injury [23]. CXCL13 has been shown to contribute to orofacial neuropathic pian via CXCR5/ERK-mediated proinflammatory cytokine production [23] as well as via activation of p38 MAPK in the trigeminal ganglion of mice [24]. Furthermore, the findings from infraorbital nerve injury studies in rats support a direct role for CXCL2 chemokine in trigeminal pain [25]. Inhibition of this cytokine results in the attenuation of mechanical allodynia to levels similar to sham-treated animals [25]. CXCL10 and its receptor CXCR3 is and additional chemokine that has been suggested to have a role in pain. It has been shown that CXCL10 acts on CXCR3 to induce ERK and AKT activation in TG neurons and contributes to the maintenance of trigeminal neuropathic pain in mice [26]. These findings suggest that pronociceptive effects of CXCL10 could act through ERK and AKT signaling.
4. Peripheral Sensitization
The state of heightened sensitivity in the peripheral tissues is termed as peripheral sensitization and is reflected by a reduction in activation threshold and elevation of nociceptive response [27]. Hyperalgesia (increased pain response to a noxious stimulus) and allodynia (pain response to non-painful stimuli) are among the characteristic features of peripheral sensitization. Some of the common clinical findings in PTNP can be explained by phenomenon of peripheral sensitization. It has been demonstrated that peripheral sensitization is associated with release of inflammatory mediators such as TNF-alpha, IL6, and IL-1 beta, alterations in receptors such as TRPV1, NGF, P2X, P2Y and neurokinin, as well as the release of CGRP and glutamatergic neurochemicals [28] (Figure 1).
5. Spontaneous Neural Activity
When a portion of the axon or cell body become sufficiently hyperexcitable, it can generate a spontaneous activity (SA) along the nociceptive path at level of axon and/or cell bodies in the sensory ganglia [29][30][31][32][33]. This process is called ectopic activity and is reported to be of two different types, Type 1 SA in the presence of subthreshold membrane potential oscillation and Type 2 in the absence of subthreshold membrane potential oscillation [29]. Within the ganglia, there are two types of conductance: a voltage-dependent, physically activating sodium conductance; and a passive, voltage-independent conductance due to potassium leak. Ectopic firing occurs when the conductance creates membrane potential oscillations and when oscillation sinusoids reach threshold amplitude [34][35][36]. These features of nerve injury may contribute to spontaneous pain seen in patients with PTNP. Inflammation around a nerve with no axonal nerve damage can become a source of pain by elevating spontaneous activity and inducing mechanosensitivity [37][38][39]. If inflammation persists, secondary nerve damage may ensue.
6. SA in Different Nerve Fibers
Animal as well as clinical electrophysiological studies demonstrate that during SA associated with painful neuropathies, an alteration of firing properties of Aβ, Aδ and C fibers occurs [31][40][41][42][43]. Spontaneous burning or sharp pain may arise from the spontaneous activity in C and Aδ fibers, whereas spontaneous activity in Aβ fibers would drive paresthesia and dysesthesia commonly present in neuropathies [44]. Animal studies show that the bulk of spontaneous activity during onset of pain is due to the spontaneous activity in Aβ fibers, and that there is a direct contribution in spontaneous and evoked pain by spontaneous activity in large diameter myelinated afferents that normally signal touch and vibration [45]. In pathologic conditions, a phenotypic switch (changing electrical characteristics and the expressed neurotransmitters complement) occurs in the Aβ fibers and they become nociceptive, acting as a source of neuropathic pain [45]. Previously injured Aβ fibers, when stimulated, were shown to induce c-fos expression, [46][47][48][49]. These findings suggest that activity in injured Aβ fibers may both drive pain and trigger central sensitization.
7. Ion Channels Alterations
Nerve injury causes variations in several ion channels in particular their expression, and function. The relative depolarization of resting potential associated with SA may be driven by the enhanced persistent sodium channel currents or reduced potassium leak currents which are active around the resting potential [44]. Hyperpolarizing cationic currents (Ih) contribute to hyperexcitability of sensitized peripheral fibers. In peripheral nerve injury, the rate of firing and current density of Ih current increases in the sensory ganglia and trigeminal ganglion [50][51][52][53][54][55][56]. Mechanical allodynia and ectopic discharge following nerve injury is reportedly reduced by blocking these currents [50][57]. The number of small-diameter sensory neurons positive for T-type Ca2+ channel increases due to the inflammatory mediators, in particular bradykinin and adenosine triphosphate (ATP), released during nerve injury and inflammation [58]. These neurons regulate peripheral sensory neuron excitability resulting in an increase in voltage gated Ca2+ channel currents in sensory neurons which leads to hypersensitivity [59][60][61][62][63]. In animal models of pain, both neuropathic and inflammatory, a silencing of Cav3.2 demonstrate an anti-nociceptive effects and pharmacological inhibitors of T-type Ca2+ channels have been shown to have analgesic effects [62][64][65][66][67][68][69][70].
8. The Role of Neurovascular Interactions in Orofacial Neuropathic Pain
Peripheral and central changes induced by nerve injury are mediated by the complex interactions between multiple cell types including neurovascular units (neurons, glial cells, endothelial cells and immunocytes) that are responsible, in part, for regulating the homeostasis of the blood–nerve barrier (BNB) [71]. Infraorbital nerve injury may lead to local disruption of the BNB and therefore increased vascular permeability. These changes result in increased infiltration of immunocytes and promote local neuroinflammation as indicated by a significant upregulation of immunocytes (CD3, CD11b) and innate immunity receptors (Toll-Like Receptor 2 (TLR2) and 4 (TLR4)) mRNA within the ION parenchyma [72][73] and neuroimmune interactions [74]. In rats exposed to infraorbital nerve chronic constriction injury, infraorbital nerve BNB disruption was linked to Hedgehog signaling and TLR4-mediated pathways [75]. Released harmful molecules such as IL-1B and chemokine MCP-1 [72] as well as immunocytes contribute to nerve sensitization and neuropathic pain [75]. Hedgehog signaling and its main effector Sonic Hedgehog (SHH) control and regulate a repair program essential for regeneration following nerve injury [76]. It has been suggested that the disruption of the BNB following nerve injury in rats is mediated by Hedgehog pathway inhibition, activation of the innate immunity [75][77] and a crosstalk between Wnt/β-catenin- and Sonic Hedgehog signaling pathways within endoneurial endothelial cells [78]. Therefore, nerve injury-induced neurovascular interactions may contribute to nerve sensitization, allodynia and neuropathic pain development.
9. The Possible Role of Microorganisms in Peripheral Sensitization
Bacterial infections may induce pain through direct or indirect effects on sensory neurons [79][80][81]. Microorganisms may trigger inflammatory reactions and expose primary afferents to algogenic substances that may drive phenotypic and functional changes in neurons and glia, immune and vascular cells in the peripheral and/or central nervous system [82]. For example, Lipopolysaccharide (LPS), a pro-inflammatory saccharide derived from Gram-negative bacteria, is responsible for the development of critical immune responses via LPS/TLR4 signaling [83]. This persistent, local inflammation-mediated sensitization can lead to chronic peripheral and central sensitization [84][85], which are well-known mechanisms leading to neuropathic pain development [86], in particular when followed by nerve injury. Additionally, LPS-mediated activation of TLR4 signaling can contribute to the disruption of the BNB, which was implicated in PTNP development [77]. Priming with LPS has been shown to induce an early pain-like behavior in rats. Following ION-CCI, mechanical static allodynia and spontaneous pain developed earlier and were more severe in LPS-pretreated rats compared to the control group [82]. Additionally, a significant increase in interleukin 1 beta (IL-1B), a key inflammatory cytokine, was observed in the TG of LPS-pretreated rats compared to controls, and a higher increase in inducible nitric oxide synthase (iNOS) was observed in ipsilateral subnucleus caudalis of LPS-pretreated rats compared to controls [82]. Therefore, inflammatory priming has been suggested to play a key role in the development and maintenance of PTNP implicating IL-1B/iNOS-dependent central sensitization mechanisms. Thus, a pre-existing inflammatory pain could be a risk factor for neuropathic pain development [87]. It is important to note that several studies have demonstrated unique characteristics of P Gingivalis that may play as immunomodulator and may also have palliative effect by increasing levels of anti-inflammatory cytokine IL-10 [88][89]. Local application of P gingivalis reduced pain-like behavior in rats exposed to the Brennan model of pain, and this anti-nociceptive property was at least partially mediated by an increase in IL-10 levels [89].
In summary, injuries at each anatomical level of the sensory system, from the peripheral ending through the sensory ganglia to the spinal cord and central nervous system structures, have been associated with neuropathic pain. Multiple possible sites for the generation of ectopic spontaneous activity have been reported in animal studies. These include along axonal projections and from the cell bodies within sensory ganglia [29][30][32][33][90]. Secondary to nerve injury, different types of cells, such as immune cells, glia and neurons, are affected at each of these anatomical levels, contributing to neuropathic pain development [91][92][93][94]. These cellular constituents interact with each other via gap junctions, synaptic transmission and cell-to-cell signaling [95][96][97]. Following nerve injury, the interactions between the different cellular components are enhanced, forming an excitatory microenvironment that may facilitate spontaneous activity [44]. Additionally, the phenotypic switching of Aβ fibers and their ectopic discharge contribute to neuropathic pain and the sensory changes associated with it. Additionally, nerve injury induced neurovascular interactions and blood–nerve-barrier disruption may contribute to nerve sensitization, allodynia and neuropathic pain development. Finally, bacteria-mediated or pre-existing inflammation may play a key role in the development and maintenance of PTNP.
10. The Role of Oxytocin in the Pathophysiology of PTNP
Oxytocin (OXT) is a neuropeptide hormone synthesized and secreted by hypothalamic neurons. Direct administration of OXT into the TG attenuated orofacial mechanical hypersensitivity following trigeminal nerve injury [98]. OXT treatment was shown to significantly increase Ik current in the TG neurons under neuropathic conditions, suggesting that OXT may promote the functional or expressional recovery of potassium channels involved in hypersensitivity induced by nerve injury [98]. Additionally, coadministration of vasopressin-1A receptor (V1A-R) antagonist entirely abolished the analgesic effects of OXT on nerve injury-induced hypersensitivity [98]. These findings suggest that OXT could interact with V1A-R, which is upregulated following nerve injury and with certain K+ channels, which are also functionally changed after nerve injury to exert its analgesic effects. Results from yet another study suggested that OXT application to the nerve-injured site attenuated ION injury-induced mechanical hypersensitivity by inhibition of the TRPV1 and TRPV4 expression in neurons innervating the whisker pad skin [99]. Finally, it has been shown that administration of oxytocin into rats’ TG-activated oxytocin receptors, attenuated orofacial ectopic pain and inhibited the upregulation of OXTR, calcitonin gene-related peptide (CGRP), IL-1B and TNFa in the TG as well as at the spinal trigeminal nucleus caudalis (SpVc) of rats exposed to inferior alveolar nerve transection [100]. These findings suggest that oxytocin may exert analgesic effect by suppressing the inflammatory response in the TG and SpVc of rats exposed to trigeminal nerve injury.
11. The Role of MicroRNAs in Pathophysiology of PTNP
MicroRNAs (miRNAs) are a class of small noncoding RNAs (approximately 22 nucleotides in length) that participate in RNA silencing and post-transcriptionally regulate gene expression. They play key roles in physiological and pathological processes [101]. MicroRNAs (miRs) are increasingly recognized as potential regulators of neuropathic pain including trigeminal neuropathic pain as they significantly regulate both immune and neuronal processes [102]. Expression levels of miR-146b-5p, −384, 155-5p, and −132-3p are markedly higher in the serum of TN patients than in healthy controls [103][104]. Additionally, MiR-186 suppresses neuroinflammation and neuropathic pain in prosopalgia mice by negatively controlling the NLRP3 inflammasome signaling [105]. Furthermore, expression of miRNA-32-5p was shown to be downregulated in the TG in response to trigeminal nerve injury in rats and promoted the development of neuropathic pain through regulation of Cav3.2 channels [106]. On the other hand, the expression of miR-195 was increased in the caudal brain stems of rats exposed to ION-CCI and its silencing alleviated trigeminal neuropathic pain by inhibiting Sonic Hedgehog (Ssh) signaling activation [16][107]. The observed upregulation of miR-195 was linked to a decreased expression of Patched1, which is the major receptor of the Sonic Hedgehog (Shh) signaling pathway [107]. These findings suggest that miR-195 is involved in the development of nerve injury induced trigeminal neuropathic pain by targeting Patched1 in the Shh signaling pathway and therefore regulating extracellular glutamate [107]. Dysregulation of microRNAs in traumatic neuropathies has also been suggested to contribute to impairment of the blood–nerve barrier (BNB) in nerve injury. For example, expression of microRNA-21-5p (miR-21) was elevated in plasma of patients with complex regional pain syndrome and in nerves of mice exposed to nerve injury. Mice presented with BNB leakage and loss of claudin-1 in injured and spared nerves [108]. Additionally, RECK gene, a putative target of miR-21, was decreased, as was downstream Mmp9 and Tgfb was upregulated in the nerves. These findings suggest that the effect of miR-21 on pain-like behavior are exerted via the inhibition of REC, the rise in MMP9 and, ultimately, claudin-1 downregulation [108]. Therefore, miR-21 was proposed to play a major role in neuropathic pain and affects pain-like behavior through different pathways, including inflammation-independent barrier impairment. Another microRNA, mir-223-3p, was shown to alleviate trigeminal neuropathic pain in male mice exposed to ION-CCI by targeting MKNK2 to suppress MAPK/ERK signaling [109].
In summary, an emerging body of evidence supports the role of micro RNAs in the pathophysiology of nerve injury-induced trigeminal neuropathic pain such as PTNP, either through direct regulation of target genes or via inflammation-independent blood–nerve barrier impairment. Taken together, the available evidence suggests that complex reciprocal neuro-immune interactions occur at both the systemic and local levels, in which miRNAs may play an active modulating role and contribute to the pathophysiology of PTNP.
12. Ganglionic Sensitization and Phenotypic Changes
Nerve injury induced phenotypic changes in satellite glial cells (SGCs) surrounding the TG [110]. Trigeminal nerve injury or inflammation induces alterations in excitability and molecular expression of voltage-gated ion channels in the TG, which causes hyperexcitability. An influx of inflammatory mediators including pain-associated molecules in the TG neuronal soma occurs by primary afferents, causing changes in intra-ganglionic communication contributing to development and maintenance of neuropathic pain [111]. SGC surrounding the TG become activated in the presence of peripheral inflammation that leads to the enhanced production and subsequent activation of neurokinin 1 (NK1) receptors with the SP released from the TG nociceptor. Membrane depolarization of the TG nociceptors is caused by the activated SGC through the enhanced production of proinflammatory cytokines (IL-1β, TNF-α) and the activation of their receptors on the nociceptor membrane. This process is reported to contribute to central sensitization and resulting allodynia and hyperalgesia [79][112][113][114][115]. Furthermore, neighboring Aβ-TG neurons are activated by the released SP, resulting in the development of mechanical allodynia by modulating their excitability [116] (Figure 2).
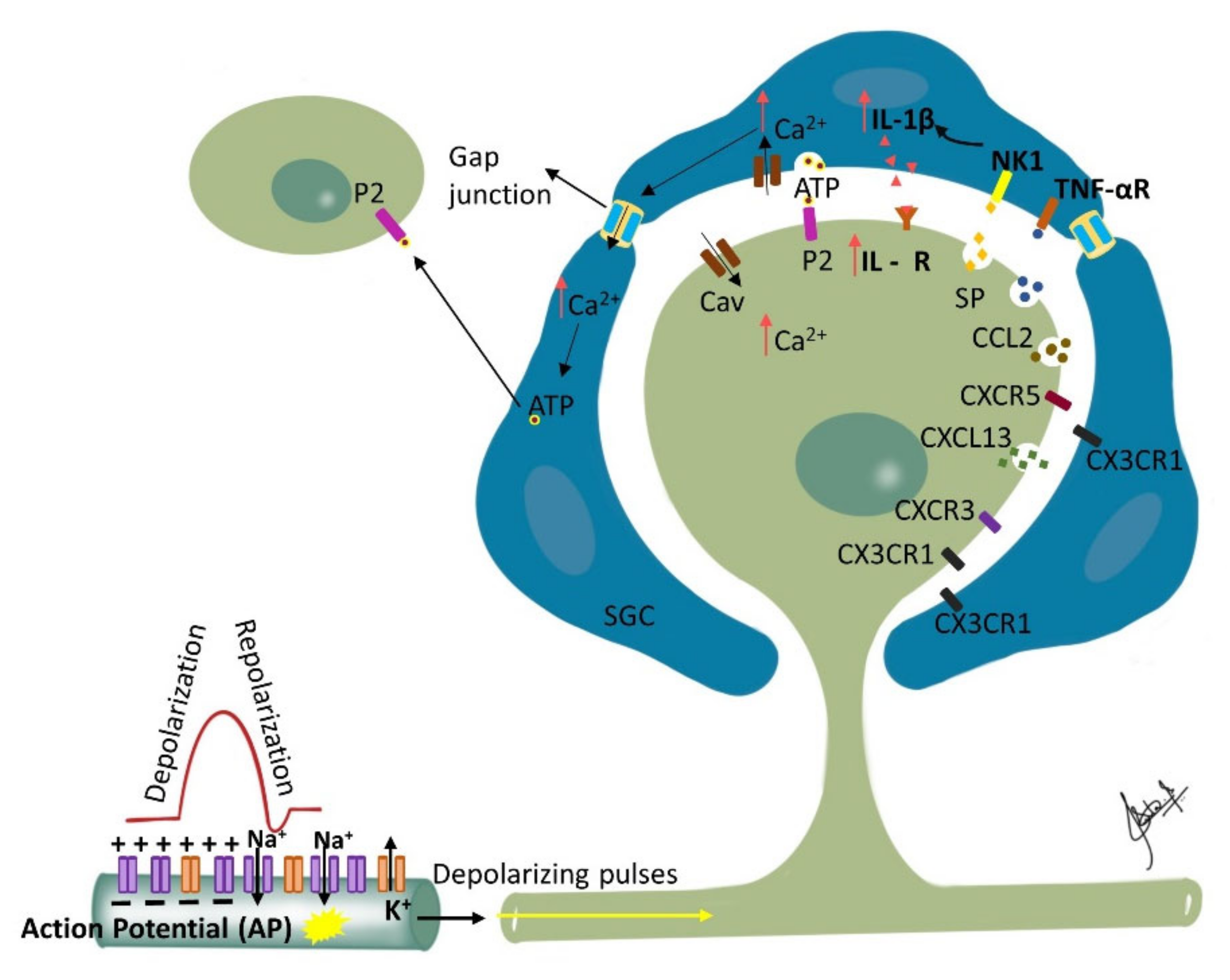
Figure 2. Ganglionic Sensitization: Changes associated with trigeminal nerve injury or inflammation lead to alterations in the cell bodies in the trigeminal ganglion (TG) that start to release pronociceptive substances. Satellite glial cells (SGC) surrounding the TG become activated in the presence of peripheral inflammation that leads to enhanced production and subsequent activation of neurokinin 1 (NK1) receptors with the SP released from the TG nociceptor. Activated SGC produce proinflammatory cytokines (IL-1β, TNF-α) that bind to their receptors on the nociceptor contributing to inflammatory hyperalgesia. Additionally, binding of the somatically released ATP to their corresponding purinergic receptors on SGC leads to increase in intracellular calcium ion concentration which propagates through gap junctions inducing release of ATP by neighboring SGC. The released ATP then binds to purinergic receptors (P2) on neighboring neurons not directly affected by the injury, and affects their excitability. Additionally, released chemokines such as CXCL13 can contribute to neuropathic pain via CXCR5/ERK mediated proinflammatory cytokine production. Furthermore, CXCL10 acts on CXCR3 to induce ERK and AKT activation in TG neurons and contributes to the maintenance of trigeminal neuropathic pain.
The change in the ganglion includes upregulation of ganglion SGCs, and up or down regulation of signaling processes/inflammatory agents/chemical mediators released in ganglion from SGCs, neuronal cells, immune cells and macrophages [28]. Proliferation of SGCs has been shown to peak at approximately 4 days post-injury and dividing SGCs were shown to preferentially surround neurons that were immunopositive for ATF-3 (a marker of nerve injury). Furthermore, nerve injury has been associated with an increase in the expression of glial fibrillary acidic protein (GFAP) in SGSs surrounding ATF-3 immunopositive and immunonegative neurons throughout the ganglia. Additionally, following nerve injury, SGCs express non-glial proteins such as CD45 and CD163, which label resident macrophages and circulating leukocytes, respectively [117]. The silencing of SGC-specific molecules such as Connexin 43, which is involved in intercellular transport or glutamate synthase (involved in glutamate recycling), can dramatically alter nociceptive responses of normal and nerve-injured rats [110]. Finally, studies investigating glutamatergic transmission within the sensory ganglia of rats showed that the soma of primary sensory neurons release glutamate when depolarized and that the somata of primary sensory neurons as well as SGCs express functional glutamate receptors at their surface [118]. Therefore, released glutamate may function as a neuroglial transmitter within the sensory ganglia. These findings suggest that glutamatergic transmission within the ganglion could impact nociceptive thresholds and that SGCs may play an important role in the genesis or maintenance of neuropathic pain. The communication between different cells in the trigeminal ganglion, including between neurons, neurons to SGCs and between the SGCs, is responsible for modulation of neuronal excitability and may play a crucial role in the clinical manifestation of sensory dysfunctions such as ectopic pain in orofacial neuropathic pain [97][116][119][120][121]. This makes the sensory ganglion function as the first relay point of pathophysiological alterations modulating afferent signals and triggering sensitization in the second order neurons and central nervous system [114][122].
13. Phenotypic Changes and Allodynia
Alterations in the expression of neuropeptides in the TG indicate that functional modifications occur secondary to nerve injury. Due to inflammation, a phenotypic shift is seen in the TG Aβ fibers, which innervates the affected area, as they start to express SP (like C fibers) [1][3][123]. Aβ fibers that transmit innocuous stimuli in healthy tissue acquire the ability to induce nociception due to peripheral stimulation, explaining allodynia. As mentioned previously, SP released by activated TG nociceptors upregulates the expression of NK1 receptors in the medium- and large-diameter neurons in the TG, altering their excitability [124][125]. This depolarization of the resting membrane potential results in an increase in SA [126]. The released SP acts as a diffusible chemical messenger, since it is released non-synaptically, mediating excitability of non-nociceptors in TG neuron, causing mechanical allodynia [124][127].
14. Response of the Injured Trigeminal System to Stress
Neuropathic pain can be exacerbated in stress and anxiety by the hyperexcitability of sympathetic nervous system activity. Upregulation of α adrenoreceptors at the site of injury and the dorsal root ganglion (DRG) that induces sensitivity to circulating catecholamines is a potential mechanism of this phenomenon. Sympathetic fiber sprouting around the neuronal cell bodies within DRG, which augments sensory–sympathetic interactions, occurs. This phenomenon is exclusive to the spinal system and has not been reported in trigeminal nerve injury, possibly explaining relatively rare sympathetically maintained orofacial pain [128][129][130].
References
- Nitzan-Luques, A.; Devor, M.; Tal, M. Genotype-selective phenotypic switch in primary afferent neurons contributes to neuropathic pain. Pain 2011, 152, 2413–2426.
- Von Hehn, C.A.; Baron, R.; Woolf, C.J. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012, 73, 638–652.
- Nitzan-Luques, A.; Minert, A.; Devor, M.; Tal, M. Dynamic genotype-selective “phenotypic switching” of CGRP expression contributes to differential neuropathic pain phenotype. Exp. Neurol. 2013, 250, 194–204.
- Kadoi, J.; Takeda, M.; Matsumoto, S. Prostaglandin E2 potentiates the excitability of small diameter trigeminal root ganglion neurons projecting onto the superficial layer of the cervical dorsal horn in rats. Exp. Brain Res. 2007, 176, 227–236.
- Takeda, M.; Takahashi, M.; Kitagawa, J.; Kanazawa, T.; Nasu, M.; Matsumoto, S. Brain-derived neurotrophic factor enhances the excitability of small-diameter trigeminal ganglion neurons projecting to the trigeminal nucleus interpolaris/caudalis transition zone following masseter muscle inflammation. Mol. Pain 2013, 9, 49.
- Takeda, M.; Ikeda, M.; Takahashi, M.; Kanazawa, T.; Nasu, M.; Matsumoto, S. Suppression of ATP-induced excitability in rat small-diameter trigeminal ganglion neurons by activation of GABAB receptor. Brain Res. Bull. 2013, 98, 155–162.
- Beyak, M.J.; Vanner, S. Inflammation-induced hyperexcitability of nociceptive gastrointestinal DRG neurones: The role of voltage-gated ion channels. Neurogastroenterol. Motil. 2005, 17, 175–186.
- Bhave, G.; Gereau, R.W.t. Posttranslational mechanisms of peripheral sensitization. J. Neurobiol. 2004, 61, 88–106.
- Chahine, M.; O’Leary, M.E. Regulation/modulation of sensory neuron sodium channels. Handb. Exp. Pharmacol. 2014, 221, 111–135.
- Lampert, A.; Eberhardt, M.; Waxman, S.G. Altered sodium channel gating as molecular basis for pain: Contribution of activation, inactivation, and resurgent currents. Handb. Exp. Pharmacol. 2014, 221, 91–110.
- Takeda, M.; Tsuboi, Y.; Kitagawa, J.; Nakagawa, K.; Iwata, K.; Matsumoto, S. Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol. Pain 2011, 7, 5.
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; Lopez Garcia, J.A. Potassium channels in neuropathic pain: Advances, challenges, and emerging ideas. Pain 2016, 157 (Suppl. S1), S7–S14.
- Montague, K.; Malcangio, M. The therapeutic potential of targeting chemokine signalling in the treatment of chronic pain. J. Neurochem. 2017, 141, 520–531.
- Solis-Castro, O.O.; Wong, N.; Boissonade, F.M. Chemokines and Pain in the Trigeminal System. Front. Pain Res. 2021, 2, 689314.
- Zhang, Z.J.; Dong, Y.L.; Lu, Y.; Cao, S.; Zhao, Z.Q.; Gao, Y.J. Chemokine CCL2 and its receptor CCR2 in the medullary dorsal horn are involved in trigeminal neuropathic pain. J. Neuroinflamm. 2012, 9, 136.
- Lin, J.; Liu, F.; Zhang, Y.Y.; Song, N.; Liu, M.K.; Fang, X.Y.; Liao, D.Q.; Zhou, C.; Wang, H.; Shen, J.F. P2Y14 receptor is functionally expressed in satellite glial cells and mediates interleukin-1beta and chemokine CCL2 secretion. J. Cell. Physiol. 2019, 234, 21199–21210.
- Dauvergne, C.; Molet, J.; Reaux-Le Goazigo, A.; Mauborgne, A.; Melik-Parsadaniantz, S.; Boucher, Y.; Pohl, M. Implication of the chemokine CCL2 in trigeminal nociception and traumatic neuropathic orofacial pain. Eur. J. Pain 2014, 18, 360–375.
- Xie, W.; Strong, J.A.; Meij, J.T.A.; Zhang, J.M.; Yu, L. Neuropathic pain: Early spontaneous afferent activity is the trigger. Pain 2005, 116, 243–256.
- Belkouch, M.; Dansereau, M.A.; Reaux-Le Goazigo, A.; Van Steenwinckel, J.; Beaudet, N.; Chraibi, A.; Melik-Parsadaniantz, S.; Sarret, P. The chemokine CCL2 increases Nav1.8 sodium channel activity in primary sensory neurons through a Gbetagamma-dependent mechanism. J. Neurosci. 2011, 31, 18381–18390.
- Van Steenwinckel, J.; Auvynet, C.; Sapienza, A.; Reaux-Le Goazigo, A.; Combadiere, C.; Melik Parsadaniantz, S. Stromal cell-derived CCL2 drives neuropathic pain states through myeloid cell infiltration in injured nerve. Brain Behav. Immun. 2015, 45, 198–210.
- Domoto, R.; Sekiguchi, F.; Tsubota, M.; Kawabata, A. Macrophage as a Peripheral Pain Regulator. Cells 2021, 10, 1881.
- Chen, O.; Donnelly, C.R.; Ji, R.R. Regulation of pain by neuro-immune interactions between macrophages and nociceptor sensory neurons. Curr. Opin. Neurobiol. 2020, 62, 17–25.
- Zhang, Q.; Cao, D.L.; Zhang, Z.J.; Jiang, B.C.; Gao, Y.J. Chemokine CXCL13 mediates orofacial neuropathic pain via CXCR5/ERK pathway in the trigeminal ganglion of mice. J. Neuroinflamm. 2016, 13, 183.
- Zhang, Q.; Zhu, M.D.; Cao, D.L.; Bai, X.Q.; Gao, Y.J.; Wu, X.B. Chemokine CXCL13 activates p38 MAPK in the trigeminal ganglion after infraorbital nerve injury. Inflammation 2017, 40, 762–769.
- Iwasa, T.; Afroz, S.; Inoue, M.; Arakaki, R.; Oshima, M.; Raju, R.; Waskitho, A.; Inoue, M.; Baba, O.; Matsuka, Y. IL-10 and CXCL2 in trigeminal ganglia in neuropathic pain. Neurosci. Lett. 2019, 703, 132–138.
- Ju, Y.Y.; Jiang, M.; Xu, F.; Wang, D.; Ding, B.; Ma, L.J.; Wu, H. CXCL10 and CXCR3 in the Trigeminal Ganglion Contribute to Trigeminal Neuropathic Pain in Mice. J. Pain Res. 2021, 14, 41–51.
- Hucho, T.; Levine, J.D. Signaling pathways in sensitization: Toward a nociceptor cell biology. Neuron 2007, 55, 365–376.
- Sessle, B.J. Chronic Orofacial Pain: Models, Mechanisms, and Genetic and Related Environmental Influences. Int. J. Mol. Sci. 2021, 22, 7112.
- Sundt, D.; Gamper, N.; Jaffe, D.B. Spike propagation through the dorsal root ganglia in an unmyelinated sensory neuron: A modeling study. J. Neurophysiol. 2015, 114, 3140–3153.
- Ma, C.; LaMotte, R.H. Multiple sites for generation of ectopic spontaneous activity in neurons of the chronically compressed dorsal root ganglion. J. Neurosci. 2007, 27, 14059–14068.
- Wall, P.D.; Devor, M. Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain 1983, 17, 321–339.
- Liu, C.N.; Wall, P.D.; Ben-Dor, E.; Michaelis, M.; Amir, R.; Devor, M. Tactile allodynia in the absence of C-fiber activation: Altered firing properties of DRG neurons following spinal nerve injury. Pain 2000, 85, 503–521.
- Coggan, J.S.; Sejnowski, T.J.; Prescott, S.A. Cooperativity between remote sites of ectopic spiking allows afterdischarge to be initiated and maintained at different locations. J. Comput. Neurosci. 2015, 39, 17–28.
- Amir, R.; Liu, C.N.; Kocsis, J.D.; Devor, M. Oscillatory mechanism in primary sensory neurones. Brain 2002, 125, 421–435.
- Amir, R.; Michaelis, M.; Devor, M. Burst discharge in primary sensory neurons: Triggered by subthreshold oscillations, maintained by depolarizing afterpotentials. J. Neurosci. 2002, 22, 1187–1198.
- Velasco, E.; Alvarez, J.L.; Meseguer, V.M.; Gallar, J.; Talavera, K. Membrane potential instabilities in sensory neurons: Mechanisms and pathophysiological relevance. Pain 2022, 163, 64–74.
- Eliav, E.; Benoliel, R.; Tal, M. Inflammation with no axonal damage of the rat saphenous nerve trunk induces ectopic discharge and mechanosensitivity in myelinated axons. Neurosci. Lett. 2001, 311, 49–52.
- Benoliel, R.; Eliav, E.; Tal, M. Strain-dependent modification of neuropathic pain behaviour in the rat hindpaw by a priming painful trigeminal nerve injury. Pain 2002, 97, 203–212.
- Chacur, M.; Milligan, E.D.; Gazda, L.S.; Armstrong, C.; Wang, H.; Tracey, K.J.; Maier, S.F.; Watkins, L.R. A new model of sciatic inflammatory neuritis (SIN): Induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain 2001, 94, 231–244.
- Orstavik, K.; Jorum, E. Microneurographic findings of relevance to pain in patients with erythromelalgia and patients with diabetic neuropathy. Neurosci. Lett. 2010, 470, 180–184.
- Campero, M.; Serra, J.; Marchettini, P.; Ochoa, J.L. Ectopic impulse generation and autoexcitation in single myelinated afferent fibers in patients with peripheral neuropathy and positive sensory symptoms. Muscle Nerve 1998, 21, 1661–1667.
- Taha, O.; Opitz, T.; Mueller, M.; Pitsch, J.; Becker, A.; Evert, B.O.; Beck, H.; Jeub, M. Neuropathic pain in experimental autoimmune neuritis is associated with altered electrophysiological properties of nociceptive DRG neurons. Exp. Neurol. 2017, 297, 25–35.
- Donadio, V.; Liguori, R. Microneurographic recording from unmyelinated nerve fibers in neurological disorders: An update. Clin. Neurophysiol. 2015, 126, 437–445.
- North, R.Y.; Lazaro, T.T.; Dougherty, P.M. Ectopic Spontaneous Afferent Activity and Neuropathic Pain. Neurosurgery 2018, 65, 49–54.
- Devor, M. Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp. Brain Res. 2009, 196, 115–128.
- Molander, C.; Hongpaisan, J.; Persson, J.K. Distribution of c-fos expressing dorsal horn neurons after electrical stimulation of low threshold sensory fibers in the chronically injured sciatic nerve. Brain Res. 1994, 644, 74–82.
- Day, A.S.; Lue, J.H.; Sun, W.Z.; Shieh, J.Y.; Wen, C.Y. A beta-fiber intensity stimulation of chronically constricted median nerve induces c-fos expression in thalamic projection neurons of the cuneate nucleus in rats with behavioral signs of neuropathic pain. Brain Res. 2001, 895, 194–203.
- Shortland, P.; Molander, C. The time-course of abeta-evoked c-fos expression in neurons of the dorsal horn and gracile nucleus after peripheral nerve injury. Brain Res. 1998, 810, 288–293.
- Fujisawa, N.; Terayama, R.; Yamaguchi, D.; Omura, S.; Yamashiro, T.; Sugimoto, T. Fos protein-like immunoreactive neurons induced by electrical stimulation in the trigeminal sensory nuclear complex of rats with chronically injured peripheral nerve. Exp. Brain Res. 2012, 219, 191–201.
- Chaplan, S.R.; Guo, H.Q.; Lee, D.H.; Luo, L.; Liu, C.; Kuei, C.; Velumian, A.A.; Butler, M.P.; Brown, S.M.; Dubin, A.E. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J. Neurosci. 2003, 23, 1169–1178.
- Yao, H.; Donnelly, D.F.; Ma, C.; LaMotte, R.H. Upregulation of the hyperpolarization-activated cation current after chronic compression of the dorsal root ganglion. J. Neurosci. 2003, 23, 2069–2074.
- Kitagawa, J.; Takeda, M.; Suzuki, I.; Kadoi, J.; Tsuboi, Y.; Honda, K.; Matsumoto, S.; Nakagawa, H.; Tanabe, A.; Iwata, K. Mechanisms involved in modulation of trigeminal primary afferent activity in rats with peripheral mononeuropathy. Eur. J. Neurosci. 2006, 24, 1976–1986.
- Cho, H.J.; Staikopoulos, V.; Furness, J.B.; Jennings, E.A. Inflammation-induced increase in hyperpolarization-activated, cyclic nucleotide-gated channel protein in trigeminal ganglion neurons and the effect of buprenorphine. Neuroscience 2009, 162, 453–461.
- He, J.T.; Li, X.Y.; Zhao, X.; Liu, X. Hyperpolarization-activated and cyclic nucleotide-gated channel proteins as emerging new targets in neuropathic pain. Rev. Neurosci. 2019, 30, 639–649.
- Cho, Y.S.; Kim, Y.S.; Moozhayil, S.J.; Yang, E.S.; Bae, Y.C. The expression of hyperpolarization-activated cyclic nucleotide-gated channel 1 (HCN1) and HCN2 in the rat trigeminal ganglion, sensory root, and dental pulp. Neuroscience 2015, 291, 15–25.
- Weng, X.; Smith, T.; Sathish, J.; Djouhri, L. Chronic inflammatory pain is associated with increased excitability and hyperpolarization-activated current (Ih) in C- but not Adelta-nociceptors. Pain 2012, 153, 900–914.
- Lee, D.H.; Chang, L.; Sorkin, L.S.; Chaplan, S.R. Hyperpolarization-activated, cation-nonselective, cyclic nucleotide-modulated channel blockade alleviates mechanical allodynia and suppresses ectopic discharge in spinal nerve ligated rats. J. Pain 2005, 6, 417–424.
- Huang, D.; Liang, C.; Zhang, F.; Men, H.; Du, X.; Gamper, N.; Zhang, H. Inflammatory mediator bradykinin increases population of sensory neurons expressing functional T-type Ca(2+) channels. Biochem. Biophys. Res. Commun. 2016, 473, 396–402.
- Marger, F.; Gelot, A.; Alloui, A.; Matricon, J.; Ferrer, J.F.; Barrere, C.; Pizzoccaro, A.; Muller, E.; Nargeot, J.; Snutch, T.P.; et al. T-type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 11268–11273.
- Garcia-Caballero, A.; Gadotti, V.M.; Stemkowski, P.; Weiss, N.; Souza, I.A.; Hodgkinson, V.; Bladen, C.; Chen, L.; Hamid, J.; Pizzoccaro, A.; et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 2014, 83, 1144–1158.
- Jagodic, M.M.; Pathirathna, S.; Joksovic, P.M.; Lee, W.; Nelson, M.T.; Naik, A.K.; Su, P.; Jevtovic-Todorovic, V.; Todorovic, S.M. Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J. Neurophysiol. 2008, 99, 3151–3156.
- Jagodic, M.M.; Pathirathna, S.; Nelson, M.T.; Mancuso, S.; Joksovic, P.M.; Rosenberg, E.R.; Bayliss, D.A.; Jevtovic-Todorovic, V.; Todorovic, S.M. Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J. Neurosci. 2007, 27, 3305–3316.
- Kang, X.J.; Chi, Y.N.; Chen, W.; Liu, F.Y.; Cui, S.; Liao, F.F.; Cai, J.; Wan, Y. Increased expression of CaV3.2 T-type calcium channels in damaged DRG neurons contributes to neuropathic pain in rats with spared nerve injury. Mol. Pain 2018, 14, 1744806918765808.
- Todorovic, S.M.; Jevtovic-Todorovic, V. Neuropathic pain: Role for presynaptic T-type channels in nociceptive signaling. Pflug. Arch. 2013, 465, 921–927.
- Messinger, R.B.; Naik, A.K.; Jagodic, M.M.; Nelson, M.T.; Lee, W.Y.; Choe, W.J.; Orestes, P.; Latham, J.R.; Todorovic, S.M.; Jevtovic-Todorovic, V. In vivo silencing of the Ca(V)3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain 2009, 145, 184–195.
- Todorovic, S.M.; Jevtovic-Todorovic, V.; Meyenburg, A.; Mennerick, S.; Perez-Reyes, E.; Romano, C.; Olney, J.W.; Zorumski, C.F. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron 2001, 31, 75–85.
- Latham, J.R.; Pathirathna, S.; Jagodic, M.M.; Choe, W.J.; Levin, M.E.; Nelson, M.T.; Lee, W.Y.; Krishnan, K.; Covey, D.F.; Todorovic, S.M.; et al. Selective T-type calcium channel blockade alleviates hyperalgesia in ob/ob mice. Diabetes 2009, 58, 2656–2665.
- Todorovic, S.M.; Pathirathna, S.; Brimelow, B.C.; Jagodic, M.M.; Ko, S.H.; Jiang, X.; Nilsson, K.R.; Zorumski, C.F.; Covey, D.F.; Jevtovic-Todorovic, V. 5beta-reduced neuroactive steroids are novel voltage-dependent blockers of T-type Ca2+ channels in rat sensory neurons in vitro and potent peripheral analgesics in vivo. Mol. Pharmacol. 2004, 66, 1223–1235.
- Cai, S.; Gomez, K.; Moutal, A.; Khanna, R. Targeting T-type/CaV3.2 channels for chronic pain. Transl. Res. 2021, 234, 20–30.
- Montera, M.; Goins, A.; Cmarko, L.; Weiss, N.; Westlund, K.N.; Alles, S.R.A. Trigeminal neuropathic pain is alleviated by inhibition of Cav3.3 T-type calcium channels in mice. Channels 2021, 15, 31–37.
- Stanimirovic, D.B.; Friedman, A. Pathophysiology of the neurovascular unit: Disease cause or consequence? J. Cereb. Blood Flow Metab. 2012, 32, 1207–1221.
- Echeverry, S.; Shi, X.Q.; Rivest, S.; Zhang, J. Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J. Neurosci. 2011, 31, 10819–10828.
- Lim, T.K.Y.; Shi, X.Q.; Martin, H.C.; Huang, H.; Luheshi, G.; Rivest, S.; Zhang, J. Blood-nerve barrier dysfunction contributes to the generation of neuropathic pain and allows targeting of injured nerves for pain relief. Pain 2014, 155, 954–967.
- Jurga, A.M.; Rojewska, E.; Piotrowska, A.; Makuch, W.; Pilat, D.; Przewlocka, B.; Mika, J. Blockade of Toll-Like Receptors (TLR2, TLR4) Attenuates Pain and Potentiates Buprenorphine Analgesia in a Rat Neuropathic Pain Model. Neural Plast. 2016, 2016, 5238730.
- Moreau, N.; Dieb, W.; Mauborgne, A.; Bourgoin, S.; Villanueva, L.; Pohl, M.; Boucher, Y. Hedgehog Pathway-Mediated Vascular Alterations Following Trigeminal Nerve Injury. J. Dent. Res. 2017, 96, 450–457.
- Nocera, G.; Jacob, C. Mechanisms of Schwann cell plasticity involved in peripheral nerve repair after injury. Cell. Mol. Life Sci. 2020, 77, 3977–3989.
- Moreau, N.; Mauborgne, A.; Bourgoin, S.; Couraud, P.O.; Romero, I.A.; Weksler, B.B.; Villanueva, L.; Pohl, M.; Boucher, Y. Early alterations of Hedgehog signaling pathway in vascular endothelial cells after peripheral nerve injury elicit blood-nerve barrier disruption, nerve inflammation, and neuropathic pain development. Pain 2016, 157, 827–839.
- Moreau, N.; Mauborgne, A.; Couraud, P.O.; Romero, I.A.; Weksler, B.B.; Villanueva, L.; Pohl, M.; Boucher, Y. Could an endoneurial endothelial crosstalk between Wnt/beta-catenin and Sonic Hedgehog pathways underlie the early disruption of the infra-orbital blood-nerve barrier following chronic constriction injury? Mol. Pain 2017, 13, 1744806917727625.
- Ji, R.R.; Chamessian, A.; Zhang, Y.Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577.
- Yang, N.J.; Chiu, I.M. Bacterial Signaling to the Nervous System through Toxins and Metabolites. J. Mol. Biol. 2017, 429, 587–605.
- Talbot, S.; Foster, S.L.; Woolf, C.J. Neuroimmunity: Physiology and Pathology. Annu. Rev. Immunol. 2016, 34, 421–447.
- Boucher, Y.; Moreau, N.; Mauborgne, A.; Dieb, W. Lipopolysaccharide-mediated inflammatory priming potentiates painful post-traumatic trigeminal neuropathy. Physiol. Behav. 2018, 194, 497–504.
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151.
- Hendrich, J.; Alvarez, P.; Joseph, E.K.; Chen, X.; Bogen, O.; Levine, J.D. Electrophysiological correlates of hyperalgesic priming in vitro and in vivo. Pain 2013, 154, 2207–2215.
- Aley, K.O.; Levine, J.D. Different peripheral mechanisms mediate enhanced nociception in metabolic/toxic and traumatic painful peripheral neuropathies in the rat. Neuroscience 2002, 111, 389–397.
- Meacham, K.; Shepherd, A.; Mohapatra, D.P.; Haroutounian, S. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr Pain Headache Rep. 2017, 21, 28.
- Dieb, W.; Moreau, N.; Chemla, I.; Descroix, V.; Boucher, Y. Neuropathic pain in the orofacial region: The role of pain history. A retrospective study. J. Stomatol. Oral Maxillofac. Surg. 2017, 118, 147–150.
- Khan, J.; Puchimada, B.; Kadouri, D.; Zusman, T.; Javed, F.; Eliav, E. The anti-nociceptive effects of Porphyromonas gingivalis lipopolysaccharide. Arch. Oral Biol. 2019, 102, 193–198.
- Liu, R.; Desta, T.; Raptis, M.; Darveau, R.P.; Graves, D.T.P. gingivalis and E. coli lipopolysaccharides exhibit different systemic but similar local induction of inflammatory markers. J. Periodontol. 2008, 79, 1241–1247.
- Amir, R.; Kocsis, J.D.; Devor, M. Multiple interacting sites of ectopic spike electrogenesis in primary sensory neurons. J. Neurosci. 2005, 25, 2576–2585.
- Mika, J.; Zychowska, M.; Popiolek-Barczyk, K.; Rojewska, E.; Przewlocka, B. Importance of glial activation in neuropathic pain. Eur. J. Pharmacol. 2013, 716, 106–119.
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368.
- Vallejo, R.; Tilley, D.M.; Vogel, L.; Benyamin, R. The role of glia and the immune system in the development and maintenance of neuropathic pain. Pain Pract. 2010, 10, 167–184.
- Donnelly, C.R.; Andriessen, A.S.; Chen, G.; Wang, K.; Jiang, C.; Maixner, W.; Ji, R.R. Central Nervous System Targets: Glial Cell Mechanisms in Chronic Pain. Neurotherapeutics 2020, 17, 846–860.
- Wu, A.; Green, C.R.; Rupenthal, I.D.; Moalem-Taylor, G. Role of gap junctions in chronic pain. J. Neurosci. Res. 2012, 90, 337–345.
- Ji, R.R.; Strichartz, G. Cell signaling and the genesis of neuropathic pain. Sci. STKE 2004, 2004, reE14.
- Hanani, M. Intercellular communication in sensory ganglia by purinergic receptors and gap junctions: Implications for chronic pain. Brain Res. 2012, 1487, 183–191.
- Kubo, A.; Shinoda, M.; Katagiri, A.; Takeda, M.; Suzuki, T.; Asaka, J.; Yeomans, D.C.; Iwata, K. Oxytocin alleviates orofacial mechanical hypersensitivity associated with infraorbital nerve injury through vasopressin-1A receptors of the rat trigeminal ganglia. Pain 2017, 158, 649–659.
- Ando, M.; Hayashi, Y.; Hitomi, S.; Shibuta, I.; Furukawa, A.; Oto, T.; Inada, T.; Matsui, T.; Fukaya, C.; Noma, N.; et al. Oxytocin-Dependent Regulation of TRPs Expression in Trigeminal Ganglion Neurons Attenuates Orofacial Neuropathic Pain Following Infraorbital Nerve Injury in Rats. Int. J. Mol. Sci. 2020, 21, 9173.
- Huang, C.L.; Liu, F.; Zhang, Y.Y.; Lin, J.; Fu, M.; Li, Y.L.; Zhou, C.; Li, C.J.; Shen, J.F. Activation of oxytocin receptor in the trigeminal ganglion attenuates orofacial ectopic pain attributed to inferior alveolar nerve injury. J. Neurophysiol. 2021, 125, 223–231.
- Tufekci, K.U.; Meuwissen, R.L.; Genc, S. The role of microRNAs in biological processes. Methods Mol. Biol. 2014, 1107, 15–31.
- Kress, M.; Huttenhofer, A.; Landry, M.; Kuner, R.; Favereaux, A.; Greenberg, D.; Bednarik, J.; Heppenstall, P.; Kronenberg, F.; Malcangio, M.; et al. microRNAs in nociceptive circuits as predictors of future clinical applications. Front. Mol. Neurosci. 2013, 6, 33.
- International Classification of Orofacial Pain, 1st edition (ICOP). Cephalalgia 2020, 40, 129–221.
- Li, X.; Wang, D.; Zhou, J.; Yan, Y.; Chen, L. Evaluation of circulating microRNA expression in patients with trigeminal neuralgia: An observational study. Medicine 2020, 99, e22972.
- Chen, M.L.; Lin, K.; Lin, S.K. NLRP3 inflammasome signaling as an early molecular response is negatively controlled by miR-186 in CFA-induced prosopalgia mice. Braz. J. Med. Biol. Res. 2018, 51, e7602.
- Qi, R.; Cao, J.; Sun, Y.; Li, Y.; Huang, Z.; Jiang, D.; Jiang, X.H.; Snutch, T.P.; Zhang, Y.; Tao, J. Histone methylation-mediated microRNA-32-5p down-regulation in sensory neurons regulates pain behaviors via targeting Cav3.2 channels. Proc. Natl. Acad. Sci. USA 2022, 119, e2117209119.
- Wang, X.; Wang, H.; Zhang, T.; He, M.; Liang, H.; Wang, H.; Xu, L.; Chen, S.; Xu, M. Inhibition of MicroRNA-195 Alleviates Neuropathic Pain by Targeting Patched1 and Inhibiting SHH Signaling Pathway Activation. Neurochem. Res. 2019, 44, 1690–1702.
- Reinhold, A.K.; Krug, S.M.; Salvador, E.; Sauer, R.S.; Karl-Scholler, F.; Malcangio, M.; Sommer, C.; Rittner, H.L. MicroRNA-21-5p functions via RECK/MMP9 as a proalgesic regulator of the blood nerve barrier in nerve injury. Ann. N. Y. Acad. Sci. 2022, 1515, 184–195.
- Huang, B.; Guo, S.; Zhang, Y.; Lin, P.; Lin, C.; Chen, M.; Zhu, S.; Huang, L.; He, J.; Zhang, L.; et al. MiR-223-3p alleviates trigeminal neuropathic pain in the male mouse by targeting MKNK2 and MAPK/ERK signaling. Brain Behav. 2022, 12, e2634.
- Jasmin, L.; Vit, J.P.; Bhargava, A.; Ohara, P.T. Can satellite glial cells be therapeutic targets for pain control? Neuron Glia Biol. 2010, 6, 63–71.
- Goto, T.; Oh, S.B.; Takeda, M.; Shinoda, M.; Sato, T.; Gunjikake, K.K.; Iwata, K. Recent advances in basic research on the trigeminal ganglion. J. Physiol. Sci. 2016, 66, 381–386.
- Takeda, M.; Tanimoto, T.; Kadoi, J.; Nasu, M.; Takahashi, M.; Kitagawa, J.; Matsumoto, S. Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain 2007, 129, 155–166.
- Takeda, M.; Kitagawa, J.; Takahashi, M.; Matsumoto, S. Activation of interleukin-1beta receptor suppresses the voltage-gated potassium currents in the small-diameter trigeminal ganglion neurons following peripheral inflammation. Pain 2008, 139, 594–602.
- Takeda, M.; Takahashi, M.; Matsumoto, S. Contribution of the activation of satellite glia in sensory ganglia to pathological pain. Neurosci. Biobehav. Rev. 2009, 33, 784–792.
- Lu, J.; Wang, D.; Xu, J.; Zhang, H.; Yu, W. New Insights on the Role of Satellite Glial Cells. Stem Cell Rev. Rep. 2022.
- Takeda, M.; Takahashi, M.; Matsumoto, S. Contribution of activated interleukin receptors in trigeminal ganglion neurons to hyperalgesia via satellite glial interleukin-1beta paracrine mechanism. Brain Behav. Immun. 2008, 22, 1016–1023.
- Donegan, M.; Kernisant, M.; Cua, C.; Jasmin, L.; Ohara, P.T. Satellite glial cell proliferation in the trigeminal ganglia after chronic constriction injury of the infraorbital nerve. Glia 2013, 61, 2000–2008.
- Kung, L.H.; Gong, K.; Adedoyin, M.; Ng, J.; Bhargava, A.; Ohara, P.T.; Jasmin, L. Evidence for glutamate as a neuroglial transmitter within sensory ganglia. PLoS ONE 2013, 8, e68312.
- Suadicani, S.O.; Cherkas, P.S.; Zuckerman, J.; Smith, D.N.; Spray, D.C.; Hanani, M. Bidirectional calcium signaling between satellite glial cells and neurons in cultured mouse trigeminal ganglia. Neuron Glia Biol. 2010, 6, 43–51.
- Huang, L.Y.; Gu, Y.; Chen, Y. Communication between neuronal somata and satellite glial cells in sensory ganglia. Glia 2013, 61, 1571–1581.
- Fan, W.; Zhu, X.; He, Y.; Zhu, M.; Wu, Z.; Huang, F.; He, H. The role of satellite glial cells in orofacial pain. J. Neurosci. Res. 2019, 97, 393–401.
- Matsuka, Y.; Afroz, S.; Dalanon, J.C.; Iwasa, T.; Waskitho, A.; Oshima, M. The role of chemical transmitters in neuron-glia interaction and pain in sensory ganglion. Neurosci. Biobehav. Rev. 2020, 108, 393–399.
- Neumann, S.; Doubell, T.P.; Leslie, T.; Woolf, C.J. Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature 1996, 384, 360–364.
- Takeda, M.; Tanimoto, T.; Ikeda, M.; Nasu, M.; Kadoi, J.; Shima, Y.; Ohta, H.; Matsumoto, S. Temporomandibular joint inflammation potentiates the excitability of trigeminal root ganglion neurons innervating the facial skin in rats. J. Neurophysiol. 2005, 93, 2723–2738.
- Takeda, M.; Takahashi, M.; Hara, N.; Matsumoto, S. Glial cell line-derived neurotrophic factor modulates the excitability of nociceptive trigeminal ganglion neurons via a paracrine mechanism following inflammation. Brain Behav. Immun. 2013, 28, 100–107.
- Xu, G.Y.; Huang, L.Y. Peripheral inflammation sensitizes P2X receptor-mediated responses in rat dorsal root ganglion neurons. J. Neurosci. 2002, 22, 93–102.
- Lolignier, S.; Eijkelkamp, N.; Wood, J.N. Mechanical allodynia. Pflug. Arch. 2015, 467, 133–139.
- Benoliel, R.; Eliav, E.; Tal, M. No sympathetic nerve sprouting in rat trigeminal ganglion following painful and non-painful infraorbital nerve neuropathy. Neurosci. Lett. 2001, 297, 151–154.
- Bongenhielm, U.; Boissonade, F.M.; Westermark, A.; Robinson, P.P.; Fried, K. Sympathetic nerve sprouting fails to occur in the trigeminal ganglion after peripheral nerve injury in the rat. Pain 1999, 82, 283–288.
- Chung, K.; Chung, J.M. Sympathetic sprouting in the dorsal root ganglion after spinal nerve ligation: Evidence of regenerative collateral sprouting. Brain Res. 2001, 895, 204–212.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
15 Dec 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No