The ICOP (International Classification of Orofacial Pain), 1ST edition describes post-traumatic trigeminal neuropathic pain (PTNP) as “a unilateral or bilateral facial or oral pain following and caused by trauma to the trigeminal nerve(s), with other symptoms and/or clinical signs of trigeminal nerve dysfunction, and persisting or recurring for more than 3 months”. A cascade of events in the peripheral and central nervous system function is involved in the pathophysiology of pain following nerve injuries. Central and peripheral processes occur in tandem and may often be co-dependent. Due to the complexity of central mechanisms, only peripheral events contributing to the pathophysiology have been reviewed herein.
- neuropathic pain
- trigeminal nerve injury
- pathophysiology
- trauma
1. Introduction
2. The Role of Receptors and Inflammatory Soup
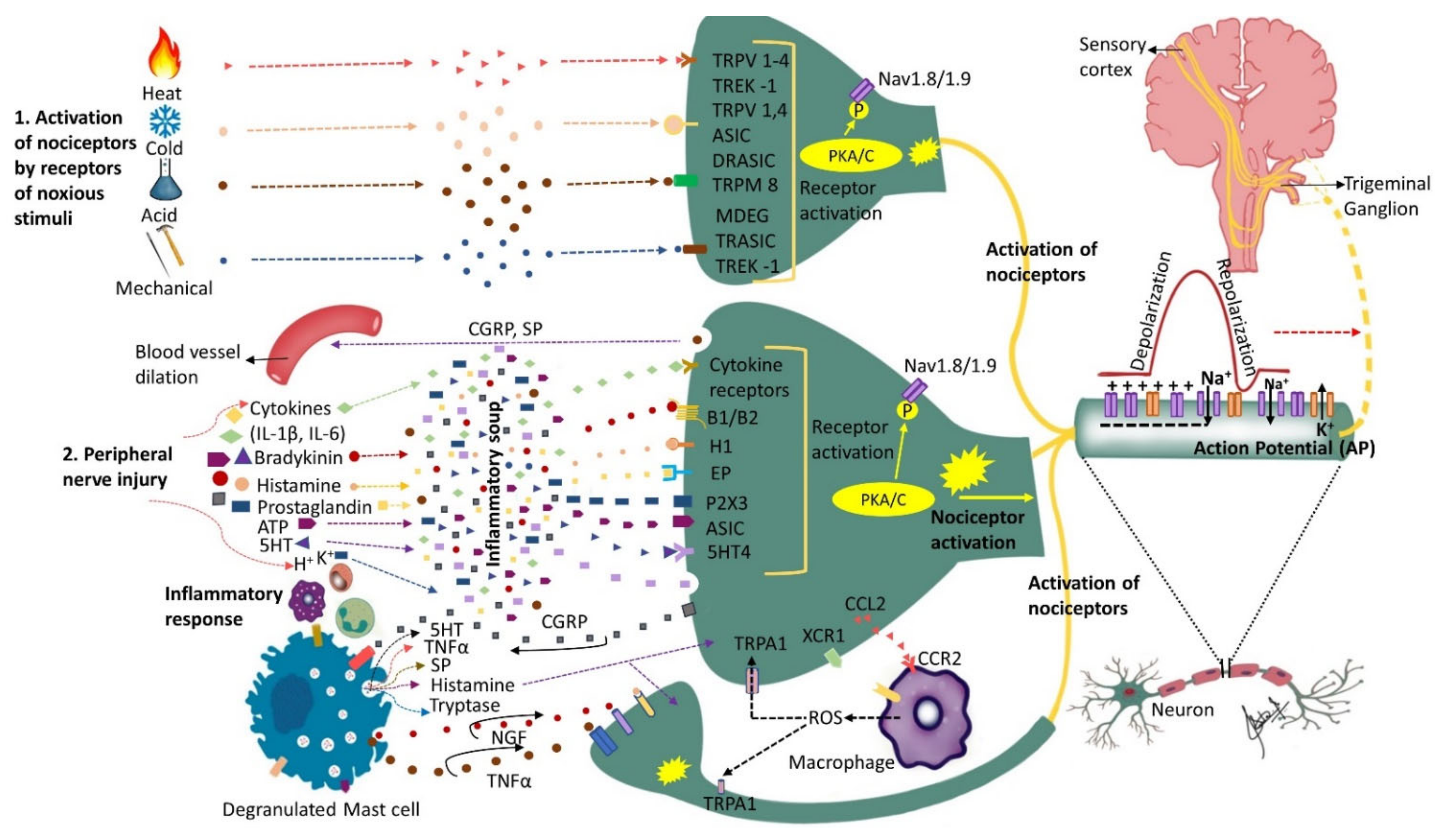
3. The Role of Chemokines
4. Peripheral Sensitization
5. Spontaneous Neural Activity
6. SA in Different Nerve Fibers
7. Ion Channels Alterations
8. The Role of Neurovascular Interactions in Orofacial Neuropathic Pain
9. The Possible Role of Microorganisms in Peripheral Sensitization
10. The Role of Oxytocin in the Pathophysiology of PTNP
11. The Role of MicroRNAs in Pathophysiology of PTNP
12. Ganglionic Sensitization and Phenotypic Changes
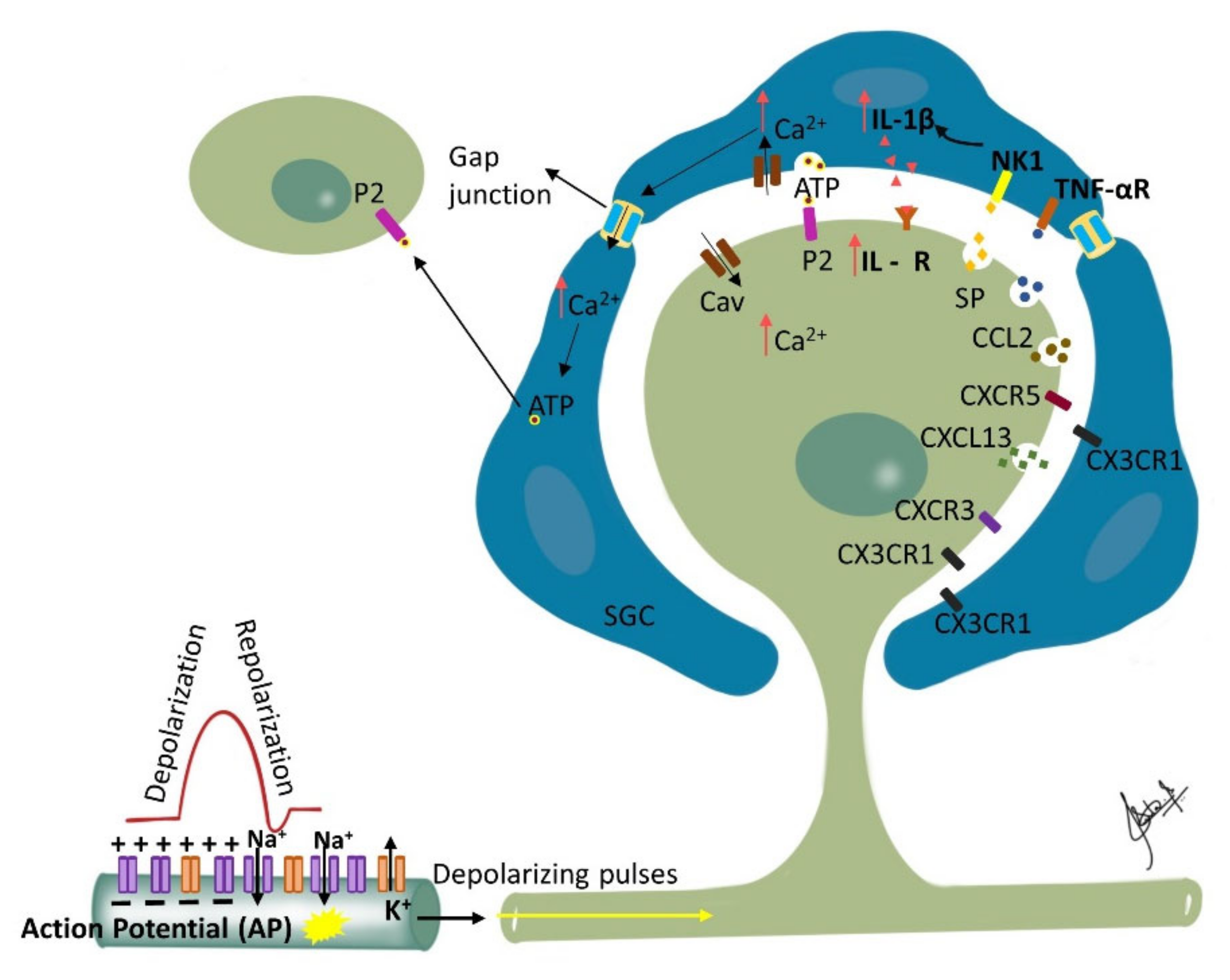
13. Phenotypic Changes and Allodynia
14. Response of the Injured Trigeminal System to Stress
This entry is adapted from the peer-reviewed paper 10.3390/biom12121753
References
- Nitzan-Luques, A.; Devor, M.; Tal, M. Genotype-selective phenotypic switch in primary afferent neurons contributes to neuropathic pain. Pain 2011, 152, 2413–2426.
- Von Hehn, C.A.; Baron, R.; Woolf, C.J. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012, 73, 638–652.
- Nitzan-Luques, A.; Minert, A.; Devor, M.; Tal, M. Dynamic genotype-selective “phenotypic switching” of CGRP expression contributes to differential neuropathic pain phenotype. Exp. Neurol. 2013, 250, 194–204.
- Kadoi, J.; Takeda, M.; Matsumoto, S. Prostaglandin E2 potentiates the excitability of small diameter trigeminal root ganglion neurons projecting onto the superficial layer of the cervical dorsal horn in rats. Exp. Brain Res. 2007, 176, 227–236.
- Takeda, M.; Takahashi, M.; Kitagawa, J.; Kanazawa, T.; Nasu, M.; Matsumoto, S. Brain-derived neurotrophic factor enhances the excitability of small-diameter trigeminal ganglion neurons projecting to the trigeminal nucleus interpolaris/caudalis transition zone following masseter muscle inflammation. Mol. Pain 2013, 9, 49.
- Takeda, M.; Ikeda, M.; Takahashi, M.; Kanazawa, T.; Nasu, M.; Matsumoto, S. Suppression of ATP-induced excitability in rat small-diameter trigeminal ganglion neurons by activation of GABAB receptor. Brain Res. Bull. 2013, 98, 155–162.
- Beyak, M.J.; Vanner, S. Inflammation-induced hyperexcitability of nociceptive gastrointestinal DRG neurones: The role of voltage-gated ion channels. Neurogastroenterol. Motil. 2005, 17, 175–186.
- Bhave, G.; Gereau, R.W.t. Posttranslational mechanisms of peripheral sensitization. J. Neurobiol. 2004, 61, 88–106.
- Chahine, M.; O’Leary, M.E. Regulation/modulation of sensory neuron sodium channels. Handb. Exp. Pharmacol. 2014, 221, 111–135.
- Lampert, A.; Eberhardt, M.; Waxman, S.G. Altered sodium channel gating as molecular basis for pain: Contribution of activation, inactivation, and resurgent currents. Handb. Exp. Pharmacol. 2014, 221, 91–110.
- Takeda, M.; Tsuboi, Y.; Kitagawa, J.; Nakagawa, K.; Iwata, K.; Matsumoto, S. Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol. Pain 2011, 7, 5.
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; Lopez Garcia, J.A. Potassium channels in neuropathic pain: Advances, challenges, and emerging ideas. Pain 2016, 157 (Suppl. S1), S7–S14.
- Montague, K.; Malcangio, M. The therapeutic potential of targeting chemokine signalling in the treatment of chronic pain. J. Neurochem. 2017, 141, 520–531.
- Solis-Castro, O.O.; Wong, N.; Boissonade, F.M. Chemokines and Pain in the Trigeminal System. Front. Pain Res. 2021, 2, 689314.
- Zhang, Z.J.; Dong, Y.L.; Lu, Y.; Cao, S.; Zhao, Z.Q.; Gao, Y.J. Chemokine CCL2 and its receptor CCR2 in the medullary dorsal horn are involved in trigeminal neuropathic pain. J. Neuroinflamm. 2012, 9, 136.
- Lin, J.; Liu, F.; Zhang, Y.Y.; Song, N.; Liu, M.K.; Fang, X.Y.; Liao, D.Q.; Zhou, C.; Wang, H.; Shen, J.F. P2Y14 receptor is functionally expressed in satellite glial cells and mediates interleukin-1beta and chemokine CCL2 secretion. J. Cell. Physiol. 2019, 234, 21199–21210.
- Dauvergne, C.; Molet, J.; Reaux-Le Goazigo, A.; Mauborgne, A.; Melik-Parsadaniantz, S.; Boucher, Y.; Pohl, M. Implication of the chemokine CCL2 in trigeminal nociception and traumatic neuropathic orofacial pain. Eur. J. Pain 2014, 18, 360–375.
- Xie, W.; Strong, J.A.; Meij, J.T.A.; Zhang, J.M.; Yu, L. Neuropathic pain: Early spontaneous afferent activity is the trigger. Pain 2005, 116, 243–256.
- Belkouch, M.; Dansereau, M.A.; Reaux-Le Goazigo, A.; Van Steenwinckel, J.; Beaudet, N.; Chraibi, A.; Melik-Parsadaniantz, S.; Sarret, P. The chemokine CCL2 increases Nav1.8 sodium channel activity in primary sensory neurons through a Gbetagamma-dependent mechanism. J. Neurosci. 2011, 31, 18381–18390.
- Van Steenwinckel, J.; Auvynet, C.; Sapienza, A.; Reaux-Le Goazigo, A.; Combadiere, C.; Melik Parsadaniantz, S. Stromal cell-derived CCL2 drives neuropathic pain states through myeloid cell infiltration in injured nerve. Brain Behav. Immun. 2015, 45, 198–210.
- Domoto, R.; Sekiguchi, F.; Tsubota, M.; Kawabata, A. Macrophage as a Peripheral Pain Regulator. Cells 2021, 10, 1881.
- Chen, O.; Donnelly, C.R.; Ji, R.R. Regulation of pain by neuro-immune interactions between macrophages and nociceptor sensory neurons. Curr. Opin. Neurobiol. 2020, 62, 17–25.
- Zhang, Q.; Cao, D.L.; Zhang, Z.J.; Jiang, B.C.; Gao, Y.J. Chemokine CXCL13 mediates orofacial neuropathic pain via CXCR5/ERK pathway in the trigeminal ganglion of mice. J. Neuroinflamm. 2016, 13, 183.
- Zhang, Q.; Zhu, M.D.; Cao, D.L.; Bai, X.Q.; Gao, Y.J.; Wu, X.B. Chemokine CXCL13 activates p38 MAPK in the trigeminal ganglion after infraorbital nerve injury. Inflammation 2017, 40, 762–769.
- Iwasa, T.; Afroz, S.; Inoue, M.; Arakaki, R.; Oshima, M.; Raju, R.; Waskitho, A.; Inoue, M.; Baba, O.; Matsuka, Y. IL-10 and CXCL2 in trigeminal ganglia in neuropathic pain. Neurosci. Lett. 2019, 703, 132–138.
- Ju, Y.Y.; Jiang, M.; Xu, F.; Wang, D.; Ding, B.; Ma, L.J.; Wu, H. CXCL10 and CXCR3 in the Trigeminal Ganglion Contribute to Trigeminal Neuropathic Pain in Mice. J. Pain Res. 2021, 14, 41–51.
- Hucho, T.; Levine, J.D. Signaling pathways in sensitization: Toward a nociceptor cell biology. Neuron 2007, 55, 365–376.
- Sessle, B.J. Chronic Orofacial Pain: Models, Mechanisms, and Genetic and Related Environmental Influences. Int. J. Mol. Sci. 2021, 22, 7112.
- Sundt, D.; Gamper, N.; Jaffe, D.B. Spike propagation through the dorsal root ganglia in an unmyelinated sensory neuron: A modeling study. J. Neurophysiol. 2015, 114, 3140–3153.
- Ma, C.; LaMotte, R.H. Multiple sites for generation of ectopic spontaneous activity in neurons of the chronically compressed dorsal root ganglion. J. Neurosci. 2007, 27, 14059–14068.
- Wall, P.D.; Devor, M. Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain 1983, 17, 321–339.
- Liu, C.N.; Wall, P.D.; Ben-Dor, E.; Michaelis, M.; Amir, R.; Devor, M. Tactile allodynia in the absence of C-fiber activation: Altered firing properties of DRG neurons following spinal nerve injury. Pain 2000, 85, 503–521.
- Coggan, J.S.; Sejnowski, T.J.; Prescott, S.A. Cooperativity between remote sites of ectopic spiking allows afterdischarge to be initiated and maintained at different locations. J. Comput. Neurosci. 2015, 39, 17–28.
- Amir, R.; Liu, C.N.; Kocsis, J.D.; Devor, M. Oscillatory mechanism in primary sensory neurones. Brain 2002, 125, 421–435.
- Amir, R.; Michaelis, M.; Devor, M. Burst discharge in primary sensory neurons: Triggered by subthreshold oscillations, maintained by depolarizing afterpotentials. J. Neurosci. 2002, 22, 1187–1198.
- Velasco, E.; Alvarez, J.L.; Meseguer, V.M.; Gallar, J.; Talavera, K. Membrane potential instabilities in sensory neurons: Mechanisms and pathophysiological relevance. Pain 2022, 163, 64–74.
- Eliav, E.; Benoliel, R.; Tal, M. Inflammation with no axonal damage of the rat saphenous nerve trunk induces ectopic discharge and mechanosensitivity in myelinated axons. Neurosci. Lett. 2001, 311, 49–52.
- Benoliel, R.; Eliav, E.; Tal, M. Strain-dependent modification of neuropathic pain behaviour in the rat hindpaw by a priming painful trigeminal nerve injury. Pain 2002, 97, 203–212.
- Chacur, M.; Milligan, E.D.; Gazda, L.S.; Armstrong, C.; Wang, H.; Tracey, K.J.; Maier, S.F.; Watkins, L.R. A new model of sciatic inflammatory neuritis (SIN): Induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain 2001, 94, 231–244.
- Orstavik, K.; Jorum, E. Microneurographic findings of relevance to pain in patients with erythromelalgia and patients with diabetic neuropathy. Neurosci. Lett. 2010, 470, 180–184.
- Campero, M.; Serra, J.; Marchettini, P.; Ochoa, J.L. Ectopic impulse generation and autoexcitation in single myelinated afferent fibers in patients with peripheral neuropathy and positive sensory symptoms. Muscle Nerve 1998, 21, 1661–1667.
- Taha, O.; Opitz, T.; Mueller, M.; Pitsch, J.; Becker, A.; Evert, B.O.; Beck, H.; Jeub, M. Neuropathic pain in experimental autoimmune neuritis is associated with altered electrophysiological properties of nociceptive DRG neurons. Exp. Neurol. 2017, 297, 25–35.
- Donadio, V.; Liguori, R. Microneurographic recording from unmyelinated nerve fibers in neurological disorders: An update. Clin. Neurophysiol. 2015, 126, 437–445.
- North, R.Y.; Lazaro, T.T.; Dougherty, P.M. Ectopic Spontaneous Afferent Activity and Neuropathic Pain. Neurosurgery 2018, 65, 49–54.
- Devor, M. Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp. Brain Res. 2009, 196, 115–128.
- Molander, C.; Hongpaisan, J.; Persson, J.K. Distribution of c-fos expressing dorsal horn neurons after electrical stimulation of low threshold sensory fibers in the chronically injured sciatic nerve. Brain Res. 1994, 644, 74–82.
- Day, A.S.; Lue, J.H.; Sun, W.Z.; Shieh, J.Y.; Wen, C.Y. A beta-fiber intensity stimulation of chronically constricted median nerve induces c-fos expression in thalamic projection neurons of the cuneate nucleus in rats with behavioral signs of neuropathic pain. Brain Res. 2001, 895, 194–203.
- Shortland, P.; Molander, C. The time-course of abeta-evoked c-fos expression in neurons of the dorsal horn and gracile nucleus after peripheral nerve injury. Brain Res. 1998, 810, 288–293.
- Fujisawa, N.; Terayama, R.; Yamaguchi, D.; Omura, S.; Yamashiro, T.; Sugimoto, T. Fos protein-like immunoreactive neurons induced by electrical stimulation in the trigeminal sensory nuclear complex of rats with chronically injured peripheral nerve. Exp. Brain Res. 2012, 219, 191–201.
- Chaplan, S.R.; Guo, H.Q.; Lee, D.H.; Luo, L.; Liu, C.; Kuei, C.; Velumian, A.A.; Butler, M.P.; Brown, S.M.; Dubin, A.E. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J. Neurosci. 2003, 23, 1169–1178.
- Yao, H.; Donnelly, D.F.; Ma, C.; LaMotte, R.H. Upregulation of the hyperpolarization-activated cation current after chronic compression of the dorsal root ganglion. J. Neurosci. 2003, 23, 2069–2074.
- Kitagawa, J.; Takeda, M.; Suzuki, I.; Kadoi, J.; Tsuboi, Y.; Honda, K.; Matsumoto, S.; Nakagawa, H.; Tanabe, A.; Iwata, K. Mechanisms involved in modulation of trigeminal primary afferent activity in rats with peripheral mononeuropathy. Eur. J. Neurosci. 2006, 24, 1976–1986.
- Cho, H.J.; Staikopoulos, V.; Furness, J.B.; Jennings, E.A. Inflammation-induced increase in hyperpolarization-activated, cyclic nucleotide-gated channel protein in trigeminal ganglion neurons and the effect of buprenorphine. Neuroscience 2009, 162, 453–461.
- He, J.T.; Li, X.Y.; Zhao, X.; Liu, X. Hyperpolarization-activated and cyclic nucleotide-gated channel proteins as emerging new targets in neuropathic pain. Rev. Neurosci. 2019, 30, 639–649.
- Cho, Y.S.; Kim, Y.S.; Moozhayil, S.J.; Yang, E.S.; Bae, Y.C. The expression of hyperpolarization-activated cyclic nucleotide-gated channel 1 (HCN1) and HCN2 in the rat trigeminal ganglion, sensory root, and dental pulp. Neuroscience 2015, 291, 15–25.
- Weng, X.; Smith, T.; Sathish, J.; Djouhri, L. Chronic inflammatory pain is associated with increased excitability and hyperpolarization-activated current (Ih) in C- but not Adelta-nociceptors. Pain 2012, 153, 900–914.
- Lee, D.H.; Chang, L.; Sorkin, L.S.; Chaplan, S.R. Hyperpolarization-activated, cation-nonselective, cyclic nucleotide-modulated channel blockade alleviates mechanical allodynia and suppresses ectopic discharge in spinal nerve ligated rats. J. Pain 2005, 6, 417–424.
- Huang, D.; Liang, C.; Zhang, F.; Men, H.; Du, X.; Gamper, N.; Zhang, H. Inflammatory mediator bradykinin increases population of sensory neurons expressing functional T-type Ca(2+) channels. Biochem. Biophys. Res. Commun. 2016, 473, 396–402.
- Marger, F.; Gelot, A.; Alloui, A.; Matricon, J.; Ferrer, J.F.; Barrere, C.; Pizzoccaro, A.; Muller, E.; Nargeot, J.; Snutch, T.P.; et al. T-type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 11268–11273.
- Garcia-Caballero, A.; Gadotti, V.M.; Stemkowski, P.; Weiss, N.; Souza, I.A.; Hodgkinson, V.; Bladen, C.; Chen, L.; Hamid, J.; Pizzoccaro, A.; et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 2014, 83, 1144–1158.
- Jagodic, M.M.; Pathirathna, S.; Joksovic, P.M.; Lee, W.; Nelson, M.T.; Naik, A.K.; Su, P.; Jevtovic-Todorovic, V.; Todorovic, S.M. Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J. Neurophysiol. 2008, 99, 3151–3156.
- Jagodic, M.M.; Pathirathna, S.; Nelson, M.T.; Mancuso, S.; Joksovic, P.M.; Rosenberg, E.R.; Bayliss, D.A.; Jevtovic-Todorovic, V.; Todorovic, S.M. Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J. Neurosci. 2007, 27, 3305–3316.
- Kang, X.J.; Chi, Y.N.; Chen, W.; Liu, F.Y.; Cui, S.; Liao, F.F.; Cai, J.; Wan, Y. Increased expression of CaV3.2 T-type calcium channels in damaged DRG neurons contributes to neuropathic pain in rats with spared nerve injury. Mol. Pain 2018, 14, 1744806918765808.
- Todorovic, S.M.; Jevtovic-Todorovic, V. Neuropathic pain: Role for presynaptic T-type channels in nociceptive signaling. Pflug. Arch. 2013, 465, 921–927.
- Messinger, R.B.; Naik, A.K.; Jagodic, M.M.; Nelson, M.T.; Lee, W.Y.; Choe, W.J.; Orestes, P.; Latham, J.R.; Todorovic, S.M.; Jevtovic-Todorovic, V. In vivo silencing of the Ca(V)3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain 2009, 145, 184–195.
- Todorovic, S.M.; Jevtovic-Todorovic, V.; Meyenburg, A.; Mennerick, S.; Perez-Reyes, E.; Romano, C.; Olney, J.W.; Zorumski, C.F. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron 2001, 31, 75–85.
- Latham, J.R.; Pathirathna, S.; Jagodic, M.M.; Choe, W.J.; Levin, M.E.; Nelson, M.T.; Lee, W.Y.; Krishnan, K.; Covey, D.F.; Todorovic, S.M.; et al. Selective T-type calcium channel blockade alleviates hyperalgesia in ob/ob mice. Diabetes 2009, 58, 2656–2665.
- Todorovic, S.M.; Pathirathna, S.; Brimelow, B.C.; Jagodic, M.M.; Ko, S.H.; Jiang, X.; Nilsson, K.R.; Zorumski, C.F.; Covey, D.F.; Jevtovic-Todorovic, V. 5beta-reduced neuroactive steroids are novel voltage-dependent blockers of T-type Ca2+ channels in rat sensory neurons in vitro and potent peripheral analgesics in vivo. Mol. Pharmacol. 2004, 66, 1223–1235.
- Cai, S.; Gomez, K.; Moutal, A.; Khanna, R. Targeting T-type/CaV3.2 channels for chronic pain. Transl. Res. 2021, 234, 20–30.
- Montera, M.; Goins, A.; Cmarko, L.; Weiss, N.; Westlund, K.N.; Alles, S.R.A. Trigeminal neuropathic pain is alleviated by inhibition of Cav3.3 T-type calcium channels in mice. Channels 2021, 15, 31–37.
- Stanimirovic, D.B.; Friedman, A. Pathophysiology of the neurovascular unit: Disease cause or consequence? J. Cereb. Blood Flow Metab. 2012, 32, 1207–1221.
- Echeverry, S.; Shi, X.Q.; Rivest, S.; Zhang, J. Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J. Neurosci. 2011, 31, 10819–10828.
- Lim, T.K.Y.; Shi, X.Q.; Martin, H.C.; Huang, H.; Luheshi, G.; Rivest, S.; Zhang, J. Blood-nerve barrier dysfunction contributes to the generation of neuropathic pain and allows targeting of injured nerves for pain relief. Pain 2014, 155, 954–967.
- Jurga, A.M.; Rojewska, E.; Piotrowska, A.; Makuch, W.; Pilat, D.; Przewlocka, B.; Mika, J. Blockade of Toll-Like Receptors (TLR2, TLR4) Attenuates Pain and Potentiates Buprenorphine Analgesia in a Rat Neuropathic Pain Model. Neural Plast. 2016, 2016, 5238730.
- Moreau, N.; Dieb, W.; Mauborgne, A.; Bourgoin, S.; Villanueva, L.; Pohl, M.; Boucher, Y. Hedgehog Pathway-Mediated Vascular Alterations Following Trigeminal Nerve Injury. J. Dent. Res. 2017, 96, 450–457.
- Nocera, G.; Jacob, C. Mechanisms of Schwann cell plasticity involved in peripheral nerve repair after injury. Cell. Mol. Life Sci. 2020, 77, 3977–3989.
- Moreau, N.; Mauborgne, A.; Bourgoin, S.; Couraud, P.O.; Romero, I.A.; Weksler, B.B.; Villanueva, L.; Pohl, M.; Boucher, Y. Early alterations of Hedgehog signaling pathway in vascular endothelial cells after peripheral nerve injury elicit blood-nerve barrier disruption, nerve inflammation, and neuropathic pain development. Pain 2016, 157, 827–839.
- Moreau, N.; Mauborgne, A.; Couraud, P.O.; Romero, I.A.; Weksler, B.B.; Villanueva, L.; Pohl, M.; Boucher, Y. Could an endoneurial endothelial crosstalk between Wnt/beta-catenin and Sonic Hedgehog pathways underlie the early disruption of the infra-orbital blood-nerve barrier following chronic constriction injury? Mol. Pain 2017, 13, 1744806917727625.
- Ji, R.R.; Chamessian, A.; Zhang, Y.Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577.
- Yang, N.J.; Chiu, I.M. Bacterial Signaling to the Nervous System through Toxins and Metabolites. J. Mol. Biol. 2017, 429, 587–605.
- Talbot, S.; Foster, S.L.; Woolf, C.J. Neuroimmunity: Physiology and Pathology. Annu. Rev. Immunol. 2016, 34, 421–447.
- Boucher, Y.; Moreau, N.; Mauborgne, A.; Dieb, W. Lipopolysaccharide-mediated inflammatory priming potentiates painful post-traumatic trigeminal neuropathy. Physiol. Behav. 2018, 194, 497–504.
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151.
- Hendrich, J.; Alvarez, P.; Joseph, E.K.; Chen, X.; Bogen, O.; Levine, J.D. Electrophysiological correlates of hyperalgesic priming in vitro and in vivo. Pain 2013, 154, 2207–2215.
- Aley, K.O.; Levine, J.D. Different peripheral mechanisms mediate enhanced nociception in metabolic/toxic and traumatic painful peripheral neuropathies in the rat. Neuroscience 2002, 111, 389–397.
- Meacham, K.; Shepherd, A.; Mohapatra, D.P.; Haroutounian, S. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr Pain Headache Rep. 2017, 21, 28.
- Dieb, W.; Moreau, N.; Chemla, I.; Descroix, V.; Boucher, Y. Neuropathic pain in the orofacial region: The role of pain history. A retrospective study. J. Stomatol. Oral Maxillofac. Surg. 2017, 118, 147–150.
- Khan, J.; Puchimada, B.; Kadouri, D.; Zusman, T.; Javed, F.; Eliav, E. The anti-nociceptive effects of Porphyromonas gingivalis lipopolysaccharide. Arch. Oral Biol. 2019, 102, 193–198.
- Liu, R.; Desta, T.; Raptis, M.; Darveau, R.P.; Graves, D.T.P. gingivalis and E. coli lipopolysaccharides exhibit different systemic but similar local induction of inflammatory markers. J. Periodontol. 2008, 79, 1241–1247.
- Amir, R.; Kocsis, J.D.; Devor, M. Multiple interacting sites of ectopic spike electrogenesis in primary sensory neurons. J. Neurosci. 2005, 25, 2576–2585.
- Mika, J.; Zychowska, M.; Popiolek-Barczyk, K.; Rojewska, E.; Przewlocka, B. Importance of glial activation in neuropathic pain. Eur. J. Pharmacol. 2013, 716, 106–119.
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368.
- Vallejo, R.; Tilley, D.M.; Vogel, L.; Benyamin, R. The role of glia and the immune system in the development and maintenance of neuropathic pain. Pain Pract. 2010, 10, 167–184.
- Donnelly, C.R.; Andriessen, A.S.; Chen, G.; Wang, K.; Jiang, C.; Maixner, W.; Ji, R.R. Central Nervous System Targets: Glial Cell Mechanisms in Chronic Pain. Neurotherapeutics 2020, 17, 846–860.
- Wu, A.; Green, C.R.; Rupenthal, I.D.; Moalem-Taylor, G. Role of gap junctions in chronic pain. J. Neurosci. Res. 2012, 90, 337–345.
- Ji, R.R.; Strichartz, G. Cell signaling and the genesis of neuropathic pain. Sci. STKE 2004, 2004, reE14.
- Hanani, M. Intercellular communication in sensory ganglia by purinergic receptors and gap junctions: Implications for chronic pain. Brain Res. 2012, 1487, 183–191.
- Kubo, A.; Shinoda, M.; Katagiri, A.; Takeda, M.; Suzuki, T.; Asaka, J.; Yeomans, D.C.; Iwata, K. Oxytocin alleviates orofacial mechanical hypersensitivity associated with infraorbital nerve injury through vasopressin-1A receptors of the rat trigeminal ganglia. Pain 2017, 158, 649–659.
- Ando, M.; Hayashi, Y.; Hitomi, S.; Shibuta, I.; Furukawa, A.; Oto, T.; Inada, T.; Matsui, T.; Fukaya, C.; Noma, N.; et al. Oxytocin-Dependent Regulation of TRPs Expression in Trigeminal Ganglion Neurons Attenuates Orofacial Neuropathic Pain Following Infraorbital Nerve Injury in Rats. Int. J. Mol. Sci. 2020, 21, 9173.
- Huang, C.L.; Liu, F.; Zhang, Y.Y.; Lin, J.; Fu, M.; Li, Y.L.; Zhou, C.; Li, C.J.; Shen, J.F. Activation of oxytocin receptor in the trigeminal ganglion attenuates orofacial ectopic pain attributed to inferior alveolar nerve injury. J. Neurophysiol. 2021, 125, 223–231.
- Tufekci, K.U.; Meuwissen, R.L.; Genc, S. The role of microRNAs in biological processes. Methods Mol. Biol. 2014, 1107, 15–31.
- Kress, M.; Huttenhofer, A.; Landry, M.; Kuner, R.; Favereaux, A.; Greenberg, D.; Bednarik, J.; Heppenstall, P.; Kronenberg, F.; Malcangio, M.; et al. microRNAs in nociceptive circuits as predictors of future clinical applications. Front. Mol. Neurosci. 2013, 6, 33.
- International Classification of Orofacial Pain, 1st edition (ICOP). Cephalalgia 2020, 40, 129–221.
- Li, X.; Wang, D.; Zhou, J.; Yan, Y.; Chen, L. Evaluation of circulating microRNA expression in patients with trigeminal neuralgia: An observational study. Medicine 2020, 99, e22972.
- Chen, M.L.; Lin, K.; Lin, S.K. NLRP3 inflammasome signaling as an early molecular response is negatively controlled by miR-186 in CFA-induced prosopalgia mice. Braz. J. Med. Biol. Res. 2018, 51, e7602.
- Qi, R.; Cao, J.; Sun, Y.; Li, Y.; Huang, Z.; Jiang, D.; Jiang, X.H.; Snutch, T.P.; Zhang, Y.; Tao, J. Histone methylation-mediated microRNA-32-5p down-regulation in sensory neurons regulates pain behaviors via targeting Cav3.2 channels. Proc. Natl. Acad. Sci. USA 2022, 119, e2117209119.
- Wang, X.; Wang, H.; Zhang, T.; He, M.; Liang, H.; Wang, H.; Xu, L.; Chen, S.; Xu, M. Inhibition of MicroRNA-195 Alleviates Neuropathic Pain by Targeting Patched1 and Inhibiting SHH Signaling Pathway Activation. Neurochem. Res. 2019, 44, 1690–1702.
- Reinhold, A.K.; Krug, S.M.; Salvador, E.; Sauer, R.S.; Karl-Scholler, F.; Malcangio, M.; Sommer, C.; Rittner, H.L. MicroRNA-21-5p functions via RECK/MMP9 as a proalgesic regulator of the blood nerve barrier in nerve injury. Ann. N. Y. Acad. Sci. 2022, 1515, 184–195.
- Huang, B.; Guo, S.; Zhang, Y.; Lin, P.; Lin, C.; Chen, M.; Zhu, S.; Huang, L.; He, J.; Zhang, L.; et al. MiR-223-3p alleviates trigeminal neuropathic pain in the male mouse by targeting MKNK2 and MAPK/ERK signaling. Brain Behav. 2022, 12, e2634.
- Jasmin, L.; Vit, J.P.; Bhargava, A.; Ohara, P.T. Can satellite glial cells be therapeutic targets for pain control? Neuron Glia Biol. 2010, 6, 63–71.
- Goto, T.; Oh, S.B.; Takeda, M.; Shinoda, M.; Sato, T.; Gunjikake, K.K.; Iwata, K. Recent advances in basic research on the trigeminal ganglion. J. Physiol. Sci. 2016, 66, 381–386.
- Takeda, M.; Tanimoto, T.; Kadoi, J.; Nasu, M.; Takahashi, M.; Kitagawa, J.; Matsumoto, S. Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain 2007, 129, 155–166.
- Takeda, M.; Kitagawa, J.; Takahashi, M.; Matsumoto, S. Activation of interleukin-1beta receptor suppresses the voltage-gated potassium currents in the small-diameter trigeminal ganglion neurons following peripheral inflammation. Pain 2008, 139, 594–602.
- Takeda, M.; Takahashi, M.; Matsumoto, S. Contribution of the activation of satellite glia in sensory ganglia to pathological pain. Neurosci. Biobehav. Rev. 2009, 33, 784–792.
- Lu, J.; Wang, D.; Xu, J.; Zhang, H.; Yu, W. New Insights on the Role of Satellite Glial Cells. Stem Cell Rev. Rep. 2022.
- Takeda, M.; Takahashi, M.; Matsumoto, S. Contribution of activated interleukin receptors in trigeminal ganglion neurons to hyperalgesia via satellite glial interleukin-1beta paracrine mechanism. Brain Behav. Immun. 2008, 22, 1016–1023.
- Donegan, M.; Kernisant, M.; Cua, C.; Jasmin, L.; Ohara, P.T. Satellite glial cell proliferation in the trigeminal ganglia after chronic constriction injury of the infraorbital nerve. Glia 2013, 61, 2000–2008.
- Kung, L.H.; Gong, K.; Adedoyin, M.; Ng, J.; Bhargava, A.; Ohara, P.T.; Jasmin, L. Evidence for glutamate as a neuroglial transmitter within sensory ganglia. PLoS ONE 2013, 8, e68312.
- Suadicani, S.O.; Cherkas, P.S.; Zuckerman, J.; Smith, D.N.; Spray, D.C.; Hanani, M. Bidirectional calcium signaling between satellite glial cells and neurons in cultured mouse trigeminal ganglia. Neuron Glia Biol. 2010, 6, 43–51.
- Huang, L.Y.; Gu, Y.; Chen, Y. Communication between neuronal somata and satellite glial cells in sensory ganglia. Glia 2013, 61, 1571–1581.
- Fan, W.; Zhu, X.; He, Y.; Zhu, M.; Wu, Z.; Huang, F.; He, H. The role of satellite glial cells in orofacial pain. J. Neurosci. Res. 2019, 97, 393–401.
- Matsuka, Y.; Afroz, S.; Dalanon, J.C.; Iwasa, T.; Waskitho, A.; Oshima, M. The role of chemical transmitters in neuron-glia interaction and pain in sensory ganglion. Neurosci. Biobehav. Rev. 2020, 108, 393–399.
- Neumann, S.; Doubell, T.P.; Leslie, T.; Woolf, C.J. Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature 1996, 384, 360–364.
- Takeda, M.; Tanimoto, T.; Ikeda, M.; Nasu, M.; Kadoi, J.; Shima, Y.; Ohta, H.; Matsumoto, S. Temporomandibular joint inflammation potentiates the excitability of trigeminal root ganglion neurons innervating the facial skin in rats. J. Neurophysiol. 2005, 93, 2723–2738.
- Takeda, M.; Takahashi, M.; Hara, N.; Matsumoto, S. Glial cell line-derived neurotrophic factor modulates the excitability of nociceptive trigeminal ganglion neurons via a paracrine mechanism following inflammation. Brain Behav. Immun. 2013, 28, 100–107.
- Xu, G.Y.; Huang, L.Y. Peripheral inflammation sensitizes P2X receptor-mediated responses in rat dorsal root ganglion neurons. J. Neurosci. 2002, 22, 93–102.
- Lolignier, S.; Eijkelkamp, N.; Wood, J.N. Mechanical allodynia. Pflug. Arch. 2015, 467, 133–139.
- Benoliel, R.; Eliav, E.; Tal, M. No sympathetic nerve sprouting in rat trigeminal ganglion following painful and non-painful infraorbital nerve neuropathy. Neurosci. Lett. 2001, 297, 151–154.
- Bongenhielm, U.; Boissonade, F.M.; Westermark, A.; Robinson, P.P.; Fried, K. Sympathetic nerve sprouting fails to occur in the trigeminal ganglion after peripheral nerve injury in the rat. Pain 1999, 82, 283–288.
- Chung, K.; Chung, J.M. Sympathetic sprouting in the dorsal root ganglion after spinal nerve ligation: Evidence of regenerative collateral sprouting. Brain Res. 2001, 895, 204–212.