Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Guangyuan Wang | -- | 4667 | 2022-12-08 10:06:43 | | | |
| 2 | Amina Yu | + 9 word(s) | 4676 | 2022-12-09 01:39:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wang, G.; Du, Y.; Ma, X.; Ye, F.; Qin, Y.; Wang, Y.; Xiang, Y.; Tao, R.; Chen, T. Thermophilic Nucleic Acid Polymerases. Encyclopedia. Available online: https://encyclopedia.pub/entry/38294 (accessed on 25 July 2026).
Wang G, Du Y, Ma X, Ye F, Qin Y, Wang Y, et al. Thermophilic Nucleic Acid Polymerases. Encyclopedia. Available at: https://encyclopedia.pub/entry/38294. Accessed July 25, 2026.
Wang, Guangyuan, Yuhui Du, Xingyun Ma, Fangkai Ye, Yanjia Qin, Yangming Wang, Yuming Xiang, Rui Tao, Tingjian Chen. "Thermophilic Nucleic Acid Polymerases" Encyclopedia, https://encyclopedia.pub/entry/38294 (accessed July 25, 2026).
Wang, G., Du, Y., Ma, X., Ye, F., Qin, Y., Wang, Y., Xiang, Y., Tao, R., & Chen, T. (2022, December 08). Thermophilic Nucleic Acid Polymerases. In Encyclopedia. https://encyclopedia.pub/entry/38294
Wang, Guangyuan, et al. "Thermophilic Nucleic Acid Polymerases." Encyclopedia. Web. 08 December, 2022.
Copy Citation
Nucleic acid polymerases are enzymes that catalyze DNA or RNA synthesis, including DNA polymerases (DNAPs), RNA polymerases (RNAPs), reverse transcriptases (RTs), and RNA-dependent RNA polymerases (RdRps), which play central roles in the storage and transmission of genetic information in living organisms, and have been widely applied in molecular biology and biotechnology. Their unique activities and functions have laid the foundation of many broadly used or modern techniques, including polymerase chain reaction (PCR), DNA sequencing, and DNA information storage. Thermostability is a desired property of nucleic acid polymerases for many of their applications, especially those involving thermocycling.
thermophilic organisms
thermophilic enzymes
nucleic acid polymerases
1. Thermophilic and Hyperthermophilic Nucleic Acid Polymerases
Generally speaking, extensively investigated and applied thermophilic and hyperthermophilic nucleic acid polymerases mainly include thermostable DNA polymerases (DNAPs) and minority RNA polymerases (RNAPs) mostly from Thermus, Thermococcus, and Pyrococcus [1][2][3] . From the perspective of family classifications, the DNAPs among these enzymes are concentrated in family A and B (Figure 1).
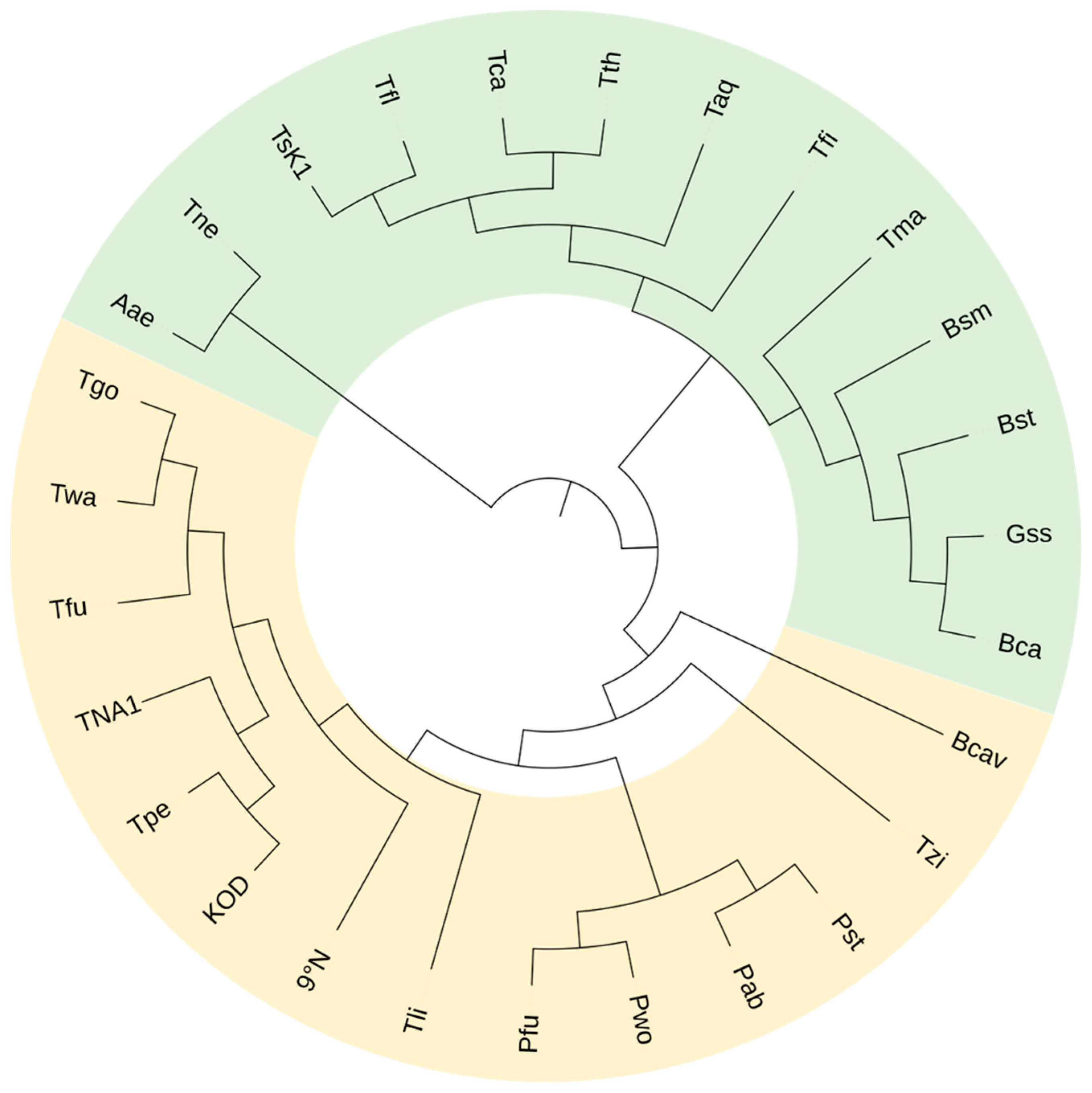
Figure 1. Phylogenetic tree of representative thermophilic DNAPs. Green: family A DNAPs; pale yellow: family B DNAPs.
Taq DNAP from the thermophilic bacterium Thermus aquaticus was the first isolated thermophilic DNAP [4], which led to a breakthrough in PCR technology by eliminating the addition of a new enzyme after each cycle of thermocycling [5][6]. The optimal temperature of Taq DNAP is 75 to 80 °C, which is much higher compared with DNAPs from organisms living in regular environments. However, its half-lives are 45 to 50 min at 95 °C and 9 min at 97.5 °C, which are relatively short [7]. Taq DNAP has been classified as family A. It has 5′-3′ exonuclease activity and no 3′-5′ exonuclease activity, so its fidelity is not good compared to polymerases that own 3′-5′ exonuclease activity [8]. Under optimized conditions, the error rate of Taq DNAP was tested to be about 1.2 × 10−5 to 3.3 × 10−6 mf × bp−1 × d−1 (mutation frequency per base pair per duplication) [9]. Based on the real-time polymerase chain reaction (PCR) experiments, amplification efficiencies of Taq DNAP were found to be around 80% for targets shorter than 1 kb and around 60% for 2.6 kb targets with a CG content between 45 to 56% [10]. Efforts have been made to alter the properties of Taq DNAP to improve its performance for different applications. For example, the pH of the reaction buffer and MgCl2 concentration have been optimized to improve its fidelity [9]. Deletion of proper regions in the 5′-3′ exonuclease domain has proven effective in improving the fidelity or thermostability of Taq DNAP. KlenTaq, a truncated variant of Taq DNAP lacking the N-terminal 235 amino acids, has been reported to have a two-fold higher fidelity than that of Taq DNAP [11]. A similar variant, the Stoffel fragment (SF), which is deficient in the N-terminal 289 amino acids, was found to have an increased thermostability [7]. Besides the DNA amplification activity, Taq DNAP also demonstrated some extent of RNA RT activity [12]. With optimized conditions, Taq DNAP could facilitate one-enzyme reverse transcription-qPCR of viral RNA [13].
Similar A-family thermophilic DNAPs have been identified from other Thermus strains, such as Tfi DNAP from Thermus filiformis [14], Tth DNAP from Thermus thermophilus [15], Tfl DNAP from Thermus flavus [16], Tca DNAP from Thermus caldophilus [17], and TsK1 DNAP from Thermus scotoductus [18]. Like Taq DNAP, these DNAPs possess 5′-3′ exonuclease activity but no 3′-5′ exonuclease activity. The PCR performances, such as amplification efficiency, fidelity, and specificity, and reaction conditions of Tfi DNAP, are similar to those of Taq DNAP [19]. Removing the 5′-3′ exonuclease domain of Tfi DNAP did not significantly affect the enzyme activity and stability [20]. However, a comparative study of exo–/exo+ Tfi DNAP blends with different blending ratios exhibited that raising the proportion of exo– Tfi mutant led to an increase in the PCR amplification yield for the target product [21]. Tth DNAP from Thermus thermophilus HB8 also showed structural and functional similarities with Taq DNAP [22]. In addition, under similar conditions in the presence of Mn2+, Tth DNAP performed higher reverse transcription activity than Taq DNAP, which is useful for one-pot reverse transcription and PCR amplification of low-level RNA [23]. Sso7d is a small protein isolated from hyperthermophilic archaebacteria Saccharolobus solfataricus and may play a role of stabilizing the genomic DNA in the cell [24][25]. It has great thermostability and is able to bind with dsDNAs without much sequence preference [26]. An early study found that the fusion of this protein with several DNAPs significantly enhanced their processivity [27]. Recently, Sso7d protein was also fused to the N-terminal of Tth DNAP, which might improve the DNA binding capacity and processivity of Tth DNAP without affecting its catalytic activity and stability [22].
Though with a high sequence homology with Taq DNAP, some family A DNAPs from other Thermus strains exhibit some different characteristics. For example, Tfl DNAP from Thermus flavus demonstrated a higher thermostability and maintained PCR activity after heat treatment at 94 °C for 60 min, while Taq DNAP lost activity within 30 min under the same temperature [28]. Furthermore, Tfl and Tth DNAPs can significantly eliminate negative influences from the inhibitors of PCR reaction in the intraocular fluids and blood, avoiding false-negative results [29][30]. For another example, Tca DNAP exhibited longer half-lives in the presence of gelatin and a narrower working pH range than that of Taq DNAP [17][31]. Recently, a novel A-family DNAP, TsK1 DNAP, was reported to have a comparable half-life to rTaq (a commercially available recombinant Taq DNAP), which is shorter than that of Taq DNAP [18]. However, this enzyme demonstrated high amplification efficiency and better fidelity than Taq DNAP, making it a potential tool for molecular biology methodologies.
Many other family A DNAPs have been isolated from Bacillus species [32], such as Bst DNAP from Bacillus stearothermophilus [33] (now categorized as Geobacillus stearothermophilus [34]), Bca DNAP from Bacillus caldotenax [35], Bcav DNAP from Bacillus caldovelox [36], Bsm DNAP from Bacillus smithii [37], and Gss DNAP from Geobacillus sp. 777 [38]. The optimal temperatures of these DNAPs are 60 to 70 °C, which are lower than those of the thermostable polymerases from Thermus species introduced above. Bst-like DNAPs are widely used in isothermal amplification techniques, such as loop-mediated isothermal amplification (LAMP) and whole genome amplification (WGA), due to their strong strand displacement activity [39][40].
Hyperthermophilic microorganisms are bacteria or archaea whose optimal temperature for growth exceeds 80 °C [41]. Thermotoga, Thermosipho, Aquifex, and Thermocrinis are common genera of hyperthermophilic bacteria [42]. Tma DNAP isolated from Thermotoga maritima is a 97 kDa A-family polymerase with inherent 3′-5′ proofreading activity and 5′-3′ exonuclease activity [43]. Tma DNAP exhibited activity over a wide range of temperatures from 45 to 90 °C, with the optimal temperature being 75 to 80 °C. N-terminal truncation of Tma DNAP yielded UlTma (Perkin-Elmer) with enhanced thermostability [44]. The presence of 3′-5′ proofreading activity does not confer a high level of fidelity to UlTma, as implied by a similar replication accuracy with that of Taq DNAP [45]. A similar polymerase, Tne DNAP, has been isolated from Thermotoga neapolitana [46]. Later research found that mutations in the O-helix region improved the fidelity of this polymerase [47]. A mixture of KlenTaq and Tne DNAPs has also been prepared and found useful for the efficient amplification of long DNA fragments [48]. Aae DNAP isolated from Aquifex aeolicus is another family A DNAP, possessing 5′-3′ polymerase activity and 3′-5′ proofreading activity but no 5′-3′ exonuclease activity [49]. Half-lives of Aae DNAP, in the presence of BSA, were 6 h and 1.7 h at 75 and 85 °C, respectively. Although Aquifex aeolicus can grow at nearly 95 °C, the activity of Aae DNAP decreased rapidly at temperatures over 90 °C.
Family B DNAPs from hyperthermophilic archaea have been widely used in PCR due to their good thermostability and 3′-5′ proofreading activity [50][51]. Several thermostable DNAPs have been isolated from the genera Thermococcus and Pyrococcus, characterized, and commercialized. Tli (Vent) DNAP from Thermococcus litoralis is an archaeal DNAP, with a molecular weight of 89 kDa [52]. It is also the first reported thermostable DNAP possessing proofreading activity, which demonstrated a 2–4 times lower error rate compared to the proofreading activity-free enzyme Replinase DNAP (isolated from Thermus flavis) [52]. Tli DNAP is extremely thermostable, having a half-life of 2 h at 100 °C, and can be used for high-temperature DNA synthesis. In addition, Tli DNAP is resistant to hemoglobin inhibition, making it suitable for PCR amplification of DNAs in blood samples [29]. KOD DNAP is another commercial high-fidelity B-family polymerase possessing a 3′-5′ exonuclease domain and was isolated from Thermococcus kodakaraensis [50] (formerly Pyrococcus sp. KOD1 [53]). KOD DNAP has a higher thermostability than most DNAPs, and its half-life at 95 °C reaches 12 h. It has also been reported to have an extension rate of 6.0–7.8 kb/min and an error rate of 2.6 × 10−6, allowing efficient and faithful amplification of DNA in PCR reaction. PCR technique based on KOD DNAP was further developed for accurate amplification of long DNAs [54]. With a mixture of wild-type KOD DNAP and its exo– variant (N210D), i.e., KOD Dash polymerase, long DNA fragments (up to 15 kb) were accurately amplified. 9°N DNAP, isolated from Thermococcus sp. 9°N-7, has a similar temperature-sensitive strand displacement activity and Km values with Tli DNAP [55]. Tgo DNAP, isolated from Thermococcus gorganarius, is another widely engineered polymerase for XNA synthesis [56]. Besides these DNAPs introduced above, many other B-family polymerases have also been isolated from Thermococcus species and characterized, including Tfu DNAP from Thermococcus fumicolans [57], TNA1 DNAP from Thermococcus sp. NA1 [58], Tpe DNAP from Thermococcus peptonophilus [59], Tzi DNAP from Thermococcus zilligii [60], and Twa DNAP from Thermococcus waiotapuensis [61].
Pfu DNAP is one of the most representative family B DNAPs isolated from hyperthermophilic marine archaea Pyrococcus furiosus [51]. Pfu DNAP has a high fidelity under optimized buffer and substrate concentrations [62]. The error rate of Pfu DNAP was found to be 1.38 × 10−6. This high fidelity is mainly attributed to the 3′-5′ exonuclease activity, and an exo– mutant of Pfu DNAP demonstrated significantly decreased fidelity [62]. The extension rate of Pfu DNAP is only 0.5–1.5 kb/min, which is lower than that of most other DNAPs [2]. The fusion of the Sso7d protein to the N-terminal of Pfu DNAP improved its processivity but did not affect its catalytic activity and stability [27]. It was also found that Pfu DNAP has weak incorporation activity of dUTP, which reduced the PCR efficiency when dUTP was used in PCR reaction [63].
Pst DNAP, also known as Deep Vent DNAP, is another well-studied thermophilic archaeal DNAP. It was isolated from Pyrococcus strain GB-D, which can grow at 104 °C [64]. Pst DNAP possesses a high fidelity with an error rate of 2.7 × 10−6, better than that of Vent or Taq DNAP [62]. Like other B-family polymerases, Pst DNAP has a high 3′-5′ proofreading activity that decreases errors during the DNA replication process. Deletion of the 3′-5′ exonuclease activity also significantly reduced its fidelity [65].
Several other thermophilic DNAPs that are not as famous as Pfu and Deep Vent DNAPs have been isolated from other Pyrococcus species. For example, Pab DNAP, which is also a B-family DNAP, was isolated from Pyrococcus abyssi, an archaeon growing in hyperthermal environments in the deep sea [66]. Pab DNAP has a higher thermostability than Taq and Pfu DNAPs and retains 75% of its activity after being incubated at 100 °C for 5 h [67]. Pab DNAP also has 3′-5′ exonuclease activity that confers proofreading ability and high fidelity to it [67]. Another example is Pwo DNAP from Pyrococcus woesei [68]. This DNAP has a molecular weight of 90 kDa, and also possesses 3′-5′ exonuclease proofreading function like other B-family DNAPs [69]. For the highest activity, Pwo DNAP needs a slightly more alkaline buffer condition, which may lead to the degradation of dNTPs, and thus dNTPs should be added right before the addition of Pwo DNAP when preparing the PCR solution [70]. Besides, the 3′-5′ exonuclease activity of this polymerase can lead to the degradation of primers and PCR products when the concentrations of dNTPs are low, and nuclease-resistant phosphorothionate protected primers can be used to solve this problem [2].
The phylogenetic tree of these polymerases shown in Figure 1 exhibits their evolutionary relationships. Although some of these DNAPs are neither well known nor commercialized, their identifications have expanded the repertoire of thermophilic DNAPs, providing more candidates to be explored and engineered for various potential applications.
Although natural thermophilic DNAPs are very efficient for DNA synthesis, and thus, have been broadly used in biotechnology, their activities of XNA synthesis are usually relatively poor, which severely limits their applications in xenobiology. In order to get efficient polymerases for the synthesis, reverse transcription, and even amplification or inter-transcription of XNAs (Figure 2), natural polymerases have to be engineered with various protein engineering strategies.
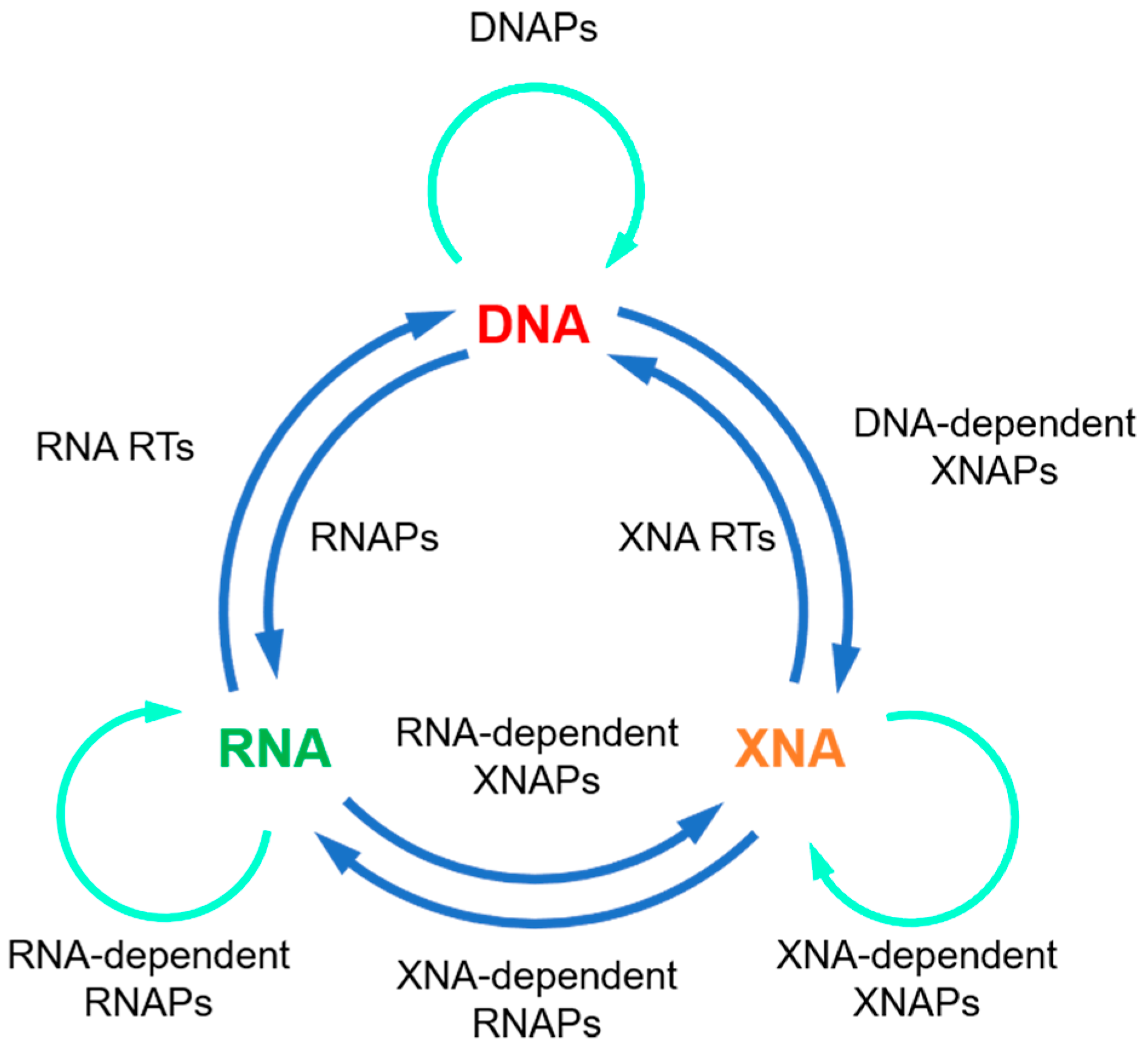
Figure 2. Expansion of the central dogma with XNAs and XNAPs. Green arrows: replication; blue arrows: transcription or reverse transcription.
2. Strategies for Engineering Thermophilic Nucleic Acid Polymerases
The engineering of polymerases can be carried out via directed evolution, rational design, or semi-rational design [1][71][72][73]. Directed evolution mimics Darwinian evolution in nature, and yet with significantly shortened evolution time for desired phenotypic traits [74]. Random mutagenesis and/or recombination are carried out on the target polymerase genes with a much higher frequency than that of spontaneous mutagenesis or recombination in nature, followed by the selection or screening of desired mutants under artificial pressures. For polymerases with more structural information, rational or semi-rational approaches can be used to predict candidate residues or regions for mutagenesis, reducing the size of the polymerase library and the labor intensity for subsequent selection or screening [75].
2.1. Strategies for Mutant Generation or Library Construction
Error-prone PCR and DNA shuffling are two methods that are most extensively employed to randomize the gene of a target protein (Figure 3). Error-prone PCR is derived from standard PCR reaction, and yet polymerases of low fidelity and altered reaction conditions, including unbalanced concentrations of dNTPs and the addition of manganese ion are applied to increase the mutation rate of the target gene during amplification [76]. DNA shuffling provides a method to recombine homologous gene sequences, which is similar to natural homologous recombination but much more efficient [77]. Many strategies have been developed for DNA shuffling, including DNase I fragmentation and reassembly, staggered extension process (StEP), and synthetic shuffling. Traditional DNA shuffling involves DNase I digestion of a pool of homologous genes and subsequent reassembly of fragments by PCR [78]. Instead of random fragmentation and assembly, StEP uses the target genes as templates to create a recombinant library through multiple rounds of shortened polymerase-catalyzed extension [79]. In some cases, the addition of specific synthetic oligonucleotides during DNA shuffling can make the libraries more directional for studying the function of interest [80]. DNA shuffling usually requires high-sequence homology. For parental genes with insufficient homology, it may be a feasible method to optimize the shuffling template sequences through computer programs to improve the homology [81][82].
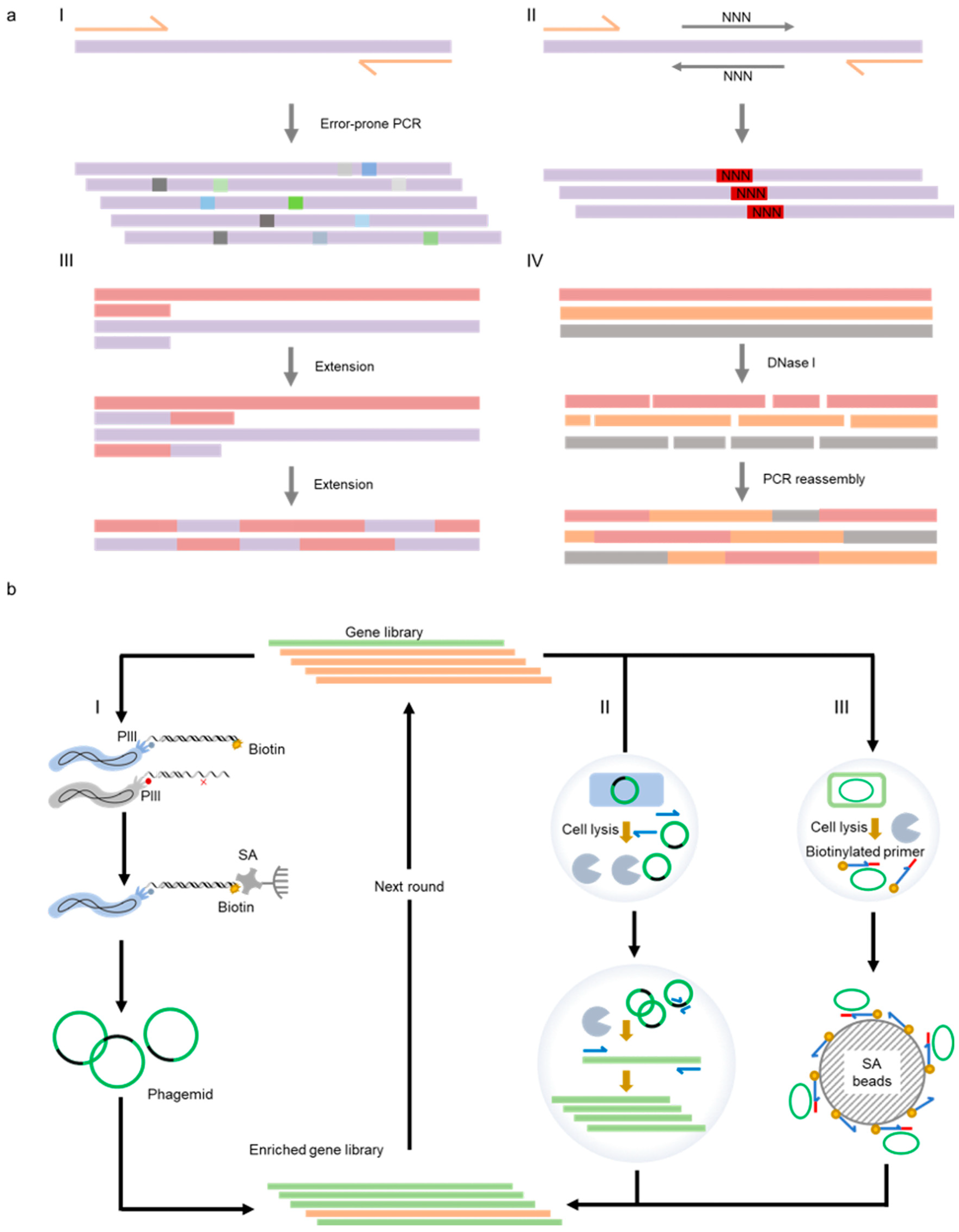
Figure 3. Representative methods employed for the generation of XNAPs. (a) Methods for the construction of polymerase libraries. I: error-prone PCR; II: site-directed saturation mutagenesis; III: gene shuffling by StEP; IV: gene shuffling by DNase I digestion and PCR reassembly; (b) methods for the selection of polymerase mutants. I: phage display; II: CSR; III: CST.
Rapid developments in sequencing, structure determination, and computational tools pave the way to the rational design of proteins. Through collection and analysis of existing sequence/structure-function data, candidate mutations of a protein for desired properties can be predicted. Site-directed mutagenesis is then carried out to generate target mutants or focused libraries. With a deeper understanding of sequence–structure-function relationships, even de novo design of proteins can be accomplished [83][84]. In past decades, many algorithms have been developed to facilitate structure prediction, design, and engineering of proteins, such as FoldX [85], Rosetta [86], I-Mutant [87], FRESCO [88], PROSS [89], and UniRep [90]. As an example of applying rational protein engineering approaches on polymerases, four Bst DNAP variants with enhanced thermostability have recently been obtained through MutCompute, an unsupervised machine learning algorithm [91].
Although having dramatically diminished the time and labor involved in the selection or screening of protein mutants, rational design requires extensive and in-depth data of sequence/structure-function relationships to improve the accuracy, which is not available for many proteins [92]. Combining the advantages of both directed evolution and rational design, semi-rational design has proven to be an effective tool for protein engineering. A small number of promising residues are identified based on computational simulation and analysis, leading to the construction of smaller but high-quality libraries and more efficient evolution processes. Various semi-rational approaches for protein engineering have been developed, including structure-based combinatorial protein engineering (SCOPE) [93], combinatorial active-site saturation test (CAST) [94], iterative saturation mutation (ISM) [95], sequence saturation mutagenesis (SeSaM) [96], protein sequence activity relationship algorithm (ProSAR) [97], and reconstructing evolutionary adaptive paths (REAP) [98]. Some of these approaches have been successfully used for the engineering of many thermophilic DNAPs, such as Bst DNAP and Taq DNAP [91][99].
2.2. Strategies for the Selection or Screening of Polymerase Libraries
Directed evolution is a powerful tool in the development of polymerases, in which the critical step is to build a high-throughput selection or screening method for the enrichment of active mutants. Selection or screening strategies for protein mutants are usually designed based on the binding of the proteins and their ligands, visualization of the catalytic activities of the enzymes, selective amplification of the target genes, or viability of the organisms [100]. The core for the selection or screening of a library is to connect the phenotypes of the mutants with their genotypes. At present, broadly used methods for selecting or screening polymerase mutants with unnatural activities mainly include methods based on the phage system, in vitro compartmentalization system, and multi-well plate system [71].
Based on the phage display technique, Romesberg and co-workers developed a polymerase selection system [99]. In this system, the polymerase library was co-displayed with the primer/template substrate on M13 phage particles, and successful extension of the primer with unnatural nucleoside triphosphates led to biotin labeling of the 3′-end of the primer, allowing the separation of active polymerase mutants from the library with streptavidin-coated beads. In another example, Liu and co-workers designed a phage-assisted continuous evolution (PACE) system, which could be used for iterative rounds of protein evolution without human intervention continuously [101]. This system correlated the desired activity of the target protein with the infectivity of the M13 phage and, thus, realized the rapid evolution of the protein along with the phage propagation.
In vitro compartmentalization is another strategy to build a linkage between genotypes and their corresponding phenotypes and has been broadly used in protein evolution. Some two decades ago, Tawfik and Griffiths developed “man-made cell-like compartments” using water-in-oil emulsions to generate separated micro-reactors, allowing the isolation of independent reactions and selection of promising protein variants [102]. Since then, emulsion-based compartmentalization has also been extensively applied to build polymerase evolution systems. Holliger and co-workers designed the compartmentalized self-replication (CSR) method based on microemulsion, in which polymerase variants were individually packaged into compartments of water-in-oil emulsion, together with PCR primers and nucleoside triphosphate substrates [103]. In this way, the genes of active variants were replicated by the polymerases that they encoded during thermocycling and enriched in the gene pool. Later, various derivative methods of CSR have been developed, including short-patch compartmentalized self-replication (spCSR) [104], reverse transcription-compartmentalized self-replication (RT-CSR) [105][106], compartmentalized partnered replication (CPR) [107], and high-temperature isothermal compartmentalized self-replication (HTI-CSR) [82].
To directly select or screen for hard-to-evolve polymerase mutants with XNA synthesis or reverse transcription activities, several other in vitro compartmentalization-based selection or screening strategies that do not rely on self-replication of the polymerase gene have been developed. For example, the compartmentalized self-tagging (CST) method was designed to select polymerases capable of XNA synthesis [108]. Similar to CSR, the polymerase pool was also compartmentalized with primers and nucleoside triphosphate substrates in water-in-oil emulsions to ensure the separation of individual variants and genotype–phenotype association. Different from CSR, CST was based on the tagging of a polymerase-encoding plasmid by extension of a short biotinylated primer when the polymerase had desired activity. Subsequent bead separation of the tagged plasmid allowed the enrichment of active polymerase variants. Later, to select for XNA RTs, Holliger and co-workers developed compartmentalized bead labeling (CBL), which relied on bead co-immobilization of the polymerase-encoding plasmids and primer/template complex for reverse transcription and subsequent fluorescent screening of the beads harboring desired variants [109].
To achieve a more controllable in vitro compartmentalization, microfluidic systems can be used to generate predefined compartments [110]. Recently, the Chaput group developed droplet-based optical polymerase sorting (DrOPS) method relying on microfluidic technology and cell sorting, in which polymerase variants were encapsulated with optical sensors for monitoring polymerase activity [111]. Successful extension of the primer by polymerase mutants led to the generation of fluorescence, and then, the water-in-oil-in-water or water-in-oil droplets were sorted by fluorescence-activated cell sorting (FACS) or fluorescence-activated droplet sorting (FADS) [112][113].
Although multi-well plate screening methods do not have throughputs as high as those of methods introduced above, they are still broadly used in the identification of polymerase variants from focused libraries with smaller size or from libraries pre-enriched with the methods introduced above [99][104][108][109]. In a typical multi-well plate screening method for polymerase mutants, the polymerase-mediated primer extension is correlated with the generation of colored or fluorescent products from enzymatic reactions, which can be directly monitored with a plate reader.
3. Thermophilic XNAPs
Thermophilic nucleic acid polymerases and their mutants have been extensively explored and used in the synthesis, reverse transcription, and even amplification of XNAs [1][114]. Although some other polymerases that are not high temperature tolerant, such as mutants of T7 RNAP, have also been used for the synthesis of modified nucleic acids [115], thermophilic nucleic acid polymerases are indispensable for XNA synthesis, reverse transcription, or amplification at high temperatures or with thermocycling programs, which are essential when tough templates with complex secondary structures are used or are important for higher yields.
Some thermophilic DNAPs, such as Taq, KlenTaq, Tth, KOD (exo–), KOD Dash, Vent (exo–), Pwo, Pfu, and Tgo DNAPs, demonstrate good tolerance to modifications on nucleobases [116][117][118][119][120][121] but are less tolerant to sugar modifications. However, syntheses or reverse transcriptions of different sugar-modified XNAs with limited lengths by certain polymerases have been reported, although not very efficient. For example, Taq DNAP was reported to be capable of reverse transcription or replication of hexose nucleic acid (HNA) with the length of a few nucleotides [122]. Bst DNAP has proven capable of reverse transcribing 2′-fluoro-arabino nucleic acid (FANA), α-L-threofuranosyl nucleic acid (TNA), and glycerol nucleic acid (GNA) [123][124]. Transcription or replication of short stretches of cyclohexenyl nucleic acid (CeNA) was demonstrated with Vent (exo–) DNAP, and reverse transcription of short CeNA was realized with Taq DNAP or Vent (exo–) DNAP [125]. Deep Vent (exo–) is able to reverse transcribe short stretches of TNA templates into DNA, and effectively incorporate all four 2′-deoxy-2′-fluoro-β-D-arabinonucleoside 5′-triphosphates (2′-F-araNTPs) on a DNA template to yield full-length FANA products [126][127]. In general, most natural polymerases show relatively narrow substrate specificities and limited activities towards XNAs. This is likely due to the fact that, in nature, to play their respective roles, polymerases have to possess stringent substrate specificities to accurately discriminate the sugars (deoxyribose and ribose) in their substrates, so that they can use the correct template (DNA or RNA) and the correct nucleoside triphosphates (dNTPs or NTPs) for the synthesis of their target products [128]. The introduction of unnatural sugars into the nucleic acids also usually leads to a great change in their structures, which may contribute to their difficult recognition by the natural polymerases as well [129]. To overcome the stringent substrate specificity and increase the activity towards unnatural substrates, natural polymerases have to be engineered.
Some commercial polymerase mutants demonstrate enhanced synthesis efficiency for modified nucleic acids. For example, Therminator DNAP, a variant of 9°N (exo–) DNAP, can incorporate various modified nucleotides [130][131][132] and even efficiently and faithfully synthesize long TNA oligonucleotides from DNA templates [132]. However, to realize efficient synthesis or reverse transcription of most of the fully substituted XNAs, the polymerases have to be further engineered via the directed evolution, rational design, or semi-rational design approaches summarized above.
Among thermophilic family A DNAPs, Taq DNAP and its truncated mutants, including SF and KlenTaq, are the most explored and engineered ones for expanded substrate repertoires. With a phage-display-based polymerase selection system, Romesberg and co-workers evolved an SF mutant, SFM19, that can incorporate 2′-O-methyl ribonucleoside triphosphates (2′-OMe-NTPs) on a DNA template [133]. Later, they further optimized the selection method and used SFM19 as the evolutionary starting point to evolve a series of polymerases, including SFM4-3, SFM4-6, and SFM4-9, that could transcribe or reverse transcribe fully 2′-OMe-modified oligonucleotides, or even PCR amplify partially 2′-OMe- or 2′-F-modified DNAs [99]. Further investigation demonstrated that these mutants could also synthesize or amplify other sugar-modified nucleic acids, including 2′-chloro (2′-Cl), 2′-amino (2′-Am), 2′-azido (2′-Az), and arabino-modified DNAs, and 2′-OMe- and 2′-F-modified RNAs [134][135][136]. Holliger and co-workers developed spCSR to select for variants of Taq DNAP, and obtained a mutant, AA40, with the ability to incorporate NTPs and sugar-modified nucleoside triphosphates [104].
Several thermophilic family B DNAPs have also been extensively engineered for the efficient synthesis and reverse transcription of various XNAs. For example, Holliger and co-workers used the CST method to select the libraries of TgoT DNAP (a Tgo DNAP mutant containing mutations V93Q, D141A, E143A, and A485L), and a mutant with HNA polymerase activity, Pol6G12, was obtained [108]. They also combined statistical correlation analysis (SCA) with activity screening or CST to develop a series of polymerases capable of synthesizing or reverse transcribing other XNAs, among which PolC7 can efficiently synthesize CeNA and LNA, PolD4K can efficiently synthesize ANA and FANA, RT521 can efficiently synthesize TNA and reverse-transcribe TNA, ANA, and FANA into DNA, while RT521K has good reverse transcription activity for CeNA and LNA. Later, by randomizing the positively charged and bulky residues of mutant RT521, which might lead to a steric clash between the polymerase surface and the P-ethyl-modification on the phosphate backbone, screening the libraries, and performing further site-directed mutagenesis, they successfully obtained mutant PGV2, which demonstrated substantially improved synthesis activity for a newly developed XNA with an uncharged backbone, alkyl phosphonate nucleic acids (phNA) [137]. Using CBL selection and plate-based screening methods, they further evolved a series of XNA RTs from mutant RT521K [109]. Among them, RT-TKK can efficiently reverse-transcribe D-altritol nucleic acid (AtNA). RT-C8, which was then evolved from RT-TKK, can efficiently reverse-transcribe 2′-OMe-RNA, and also has some extent of reverse transcription activity for P-α-S-phosphorothioate 2′-methoxyethyl RNA (PS 2′-MOE-RNA). Another derivative of RT-TKK, RT-H4, can reverse transcribe HNA much more efficiently than RT521K and RT-TKK.
Chaput and co-workers identified specificity-determining residues (SDRs) of the polymerase by analyzing the polymerase/DNA complex structure and screened for the beneficial mutations at specificity-determining residues (SDRs) positions in a model polymerase scaffold [138]. By transferring these mutations to homologous proteins, a series of mutants that demonstrated RNA and TNA synthesis activities were rapidly developed from several family B DNAPs, including 9°N, Tgo, KOD, and Deep Vent DNAPs. They also successfully selected a manganese-independent TNA polymerase, 9n-YRI, from a site-saturation mutagenesis library of 9°N DNAP with the DrOPS method that they developed [111]. They further combined FADS sorting with deep mutational scanning to provide an unbiased screening of all possible single-point mutations in the finger subdomain of KOD (exo–) DNAP [113]. By screening mutants containing combinations of selected mutations, a double mutant, KOD-RS, which can conduct efficient TNA synthesis, was obtained, suggesting that polymerase specificity may be controlled by a small number of highly specific residues and more attention should be paid to these sites when engineering polymerases for the synthesis of specific nucleic acids. They later developed a programmed allelic mutation (PAM) strategy, applied it with DrOPS sorting, and successfully evolved a mutant with enhanced efficiency and specificity for TNA synthesis, Kod-RSGA, from mutant Kod-RS [139]. Herdewijn and co-workers reported 3′-2′ phosphonomethylthreosyl nucleic acid (tPhoNA or PMT) as a novel genetic material, and carried out stepwise engineering of TgoT DNAP to produce a PMT polymerase [140]. By introducing mutations that are related to XNA synthesis activity and screening for mutations at key residues based on previously reported mutants, they successfully obtained mutant TgoT-EPFLH, which can efficiently synthesize PMT. They also demonstrated that PMT could be efficiently reverse transcribed into DNA by both TgoT mutant RT521 and KOD mutant K.RT521K. Based on structural analysis, Hoshino et al. developed variants of KOD DNAP for LNA synthesis and reverse transcription, among which KOD-DGLNK can efficiently synthesize LNA from DNA, and KOD-DLK can efficiently reverse transcribe LNA into DNA [141]. These two mutants also demonstrated transcription or reverse transcription activity for 2′-OMe-RNA, respectively. Recently, Chaput and co-workers systematically compared some of the representative XNAPs obtained in previous works introduced above and demonstrated their diversity in thermostability and activity, specificity, and fidelity for the synthesis or reverse transcription of different nucleic acids, including RNA, FANA, ANA, HNA, TNA, and PMT [142].
References
- Laos, R.; Thomson, J.M.; Benner, S.A. DNA Polymerases Engineered by Directed Evolution to Incorporate Non-Standard Nucleotides. Front. Microbiol. 2014, 5, 565.
- Terpe, K. Overview of Thermostable DNA Polymerases for Classical PCR Applications: From Molecular and Biochemical Fundamentals to Commercial Systems. Appl. Microbiol. Biotechnol. 2013, 97, 10243–10254.
- Date, T.; Suzuki, K.; Imahori, K. Purification and Some Properties of DNA-Dependent RNA Polymerase from an Extreme Thermophile, Thermus thermophilus HB8. J. Biochem. 1975, 78, 845–858.
- Chien, A.; Edgar, D.B.; Trela, J.M. Deoxyribonucleic Acid Polymerase from the Extreme Thermophile Thermus aquaticus. J. Bacteriol. 1976, 127, 1550–1557.
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific Enzymatic Amplification of DNA In Vitro: The Polymerase Chain Reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–273.
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-Directed Enzymatic Amplification of DNA with a Thermostable DNA Polymerase. Science 1988, 239, 487–491.
- Lawyer, F.C.; Stoffel, S.; Saiki, R.K.; Chang, S.Y.; Landre, P.A.; Abramson, R.D.; Gelfand, D.H. High-Level Expression, Purification, and Enzymatic Characterization of Full-Length Thermus aquaticus DNA Polymerase and a Truncated form Deficient in 5′ to 3′ Exonuclease Activity. PCR Methods Appl. 1993, 2, 275–287.
- Tindall, K.R.; Kunkel, T.A. Fidelity of DNA Synthesis by the Thermus aquaticus DNA Polymerase. Biochemistry 1988, 27, 6008–6013.
- Eckert, K.A.; Kunkel, T.A. High Fidelity DNA Synthesis by the Thermus aquaticus DNA Polymerase. Nucleic Acids Res. 1990, 18, 3739–3744.
- Arezi, B.; Xing, W.; Sorge, J.A.; Hogrefe, H.H. Amplification Efficiency of Thermostable DNA Polymerases. Anal. Biochem. 2003, 321, 226–235.
- Barnes, W.M. The Fidelity of Taq Polymerase Catalyzing PCR Is Improved by an N-Terminal Deletion. Gene 1992, 112, 29–35.
- Grabko, V.I.; Chistyakova, L.G.; Lyapustin, V.N.; Korobko, V.G.; Miroshnikov, A.I. Reverse Transcription, Amplification and Sequencing of Poliovirus RNA by Taq DNA Polymerase. FEBS Lett. 1996, 387, 189–192.
- Bhadra, S.; Maranhao, A.C.; Paik, I.; Ellington, A.D. One-Enzyme Reverse Transcription qPCR Using Taq DNA Polymerase. Biochemistry 2020, 59, 4638–4645.
- Jung, S.E.; Choi, J.J.; Kim, H.K.; Kwon, S.T. Cloning and Analysis of the DNA Polymerase-Encoding Gene from Thermus filiformis. Mol. Cells. 1997, 7, 769–776.
- Dabrowski, S.; Kur, J. Recombinant His-Tagged DNA Polymerase. I. Cloning, Purification and Partial Characterization of Thermus thermophilus Recombinant DNA Polymerase. Acta Biochim. Pol. 1998, 45, 653–660.
- Akhmetzjanov, A.A.; Vakhitov, V.A. Molecular Cloning and Nucleotide Sequence of the DNA Polymerase Gene from Thermus flavus. Nucleic Acids Res. 1992, 20, 5839.
- Park, J.H.; Kim, J.S.; Kwon, S.T.; Lee, D.S. Purification and Characterization of Thermus caldophilus GK24 DNA Polymerase. Eur. J. Biochem. 1993, 214, 135–140.
- Saghatelyan, A.; Panosyan, H.; Trchounian, A.; Birkeland, N.K. Characteristics of DNA Polymerase I from an Extreme Thermophile, Thermus scotoductus Strain K1. MicrobiologyOpen 2021, 10, e1149.
- Choi, J.J.; Jung, S.E.; Kim, H.K.; Kwon, S.T. Purification and Properties of Thermus filiformis DNA Polymerase Expressed in Escherichia coli. Biotechnol. Appl. Biochem. 1999, 30, 19–25.
- Choi, J.J.; Kim, H.K.; Kwon, S.T. Purification and Characterization of the 5′→3′ Exonuclease Domain-Deleted Thermus filiformis DNA Polymerase Expressed in Escherichia coli. Biotechnol. Lett. 2001, 23, 1647–1652.
- Zheng, W.; Lee, J.E.; Potter, R.J.; Mandelman, D. DNA Polymerase Blends and Mutant DNA Polymerases. U.S. Patent Application No 11/170,762, 28 December 2006.
- Aye, S.L.; Fujiwara, K.; Ueki, A.; Doi, N. Engineering of DNA Polymerase I from Thermus thermophilus Using Compartmentalized Self-Replication. Biochem. Biophys. Res. Commun. 2018, 499, 170–176.
- Myers, T.W.; Gelfand, D.H. Reverse Transcription and DNA Amplification by a Thermus thermophilus DNA Polymerase. Biochemistry 1991, 30, 7661–7666.
- Choli, T.; Henning, P.; Wittmann-Liebold, B.; Reinhardt, R. Isolation, Characterization and Microsequence Analysis of a Small Basic Methylated DNA-Binding Protein from the Archaebacterium, Sulfolobus solfataricus. Biochim. Biophys. Acta. 1988, 950, 193–203.
- Sakai, H.D.; Kurosawa, N. Saccharolobus caldissimus Gen. nov., sp nov., a Facultatively Anaerobic Iron-Reducing Hyperthermophilic Archaeon Isolated from an Acidic Terrestrial Hot Spring, and Reclassification of Sulfolobus solfataricus as Saccharolobus solfataricus comb. Nov and Sulfolobus shibatae as Saccharolobus shibatae comb. nov. Int. J. Syst. Evol. Micr. 2018, 68, 1271–1278.
- Gao, Y.G.; Su, S.Y.; Robinson, H.; Padmanabhan, S.; Lim, L.; McCrary, B.S.; Edmondson, S.P.; Shriver, J.W.; Wang, A.H. The Crystal Structure of the Hyperthermophile Chromosomal Protein Sso7d Bound to DNA. Nat. Struct. Biol. 1998, 5, 782–786.
- Wang, Y.; Prosen, D.E.; Mei, L.; Sullivan, J.C.; Finney, M.; Vander Horn, P.B. A Novel Strategy to Engineer DNA Polymerases for Enhanced Processivity and Improved Performance in Vitro. Nucleic Acids Res. 2004, 32, 1197–1207.
- Harrell, R.A.; Hart, R.P. Rapid Preparation of Thermus flavus DNA Polymerase. PCR Methods Appl. 1994, 3, 372–375.
- Al-Soud, W.A.; Radstrom, P. Purification and Characterization of PCR-Inhibitory Components in Blood Cells. J. Clin. Microbiol. 2001, 39, 485–493.
- Wiedbrauk, D.L.; Werner, J.C.; Drevon, A.M. Inhibition of PCR by Aqueous and Vitreous Fluids. J. Clin. Microbiol. 1995, 33, 2643–2646.
- Kwon, S.T.; Kim, J.S.; Park, J.H.; Kim, H.K.; Lee, D.S. Cloning and Analysis of the DNA Polymerase-Encoding Gene from Thermus caldophilus GK24. Mol. Cells. 1997, 7, 264–271.
- Perler, F.B.; Kumar, S.; Kong, H. Thermostable DNA Polymerases. Adv. Protein Chem. 1996, 48, 377–435.
- Aliotta, J.M.; Pelletier, J.J.; Ware, J.L.; Moran, L.S.; Benner, J.S.; Kong, H. Thermostable Bst DNA Polymerase I Lacks a 3′→5′ Proofreading Exonuclease Activity. Genet. Anal. Biomol. Eng. 1996, 12, 185–195.
- Nazina, T.N.; Tourova, T.P.; Poltaraus, A.B.; Novikova, E.V.; Grigoryan, A.A.; Ivanova, A.E.; Lysenko, A.M.; Petrunyaka, V.V.; Osipov, G.A.; Belyaev, S.S.; et al. Taxonomic Study of Aerobic Thermophilic Bacilli: Descriptions of Geobacillus subterraneus Gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from Petroleum Reservoirs and Transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermodenitrificans to Geobacillus as the New Combinations G. stearothermophilus, G. thermocatenulatus, G. thermoleovorans, G. kaustophilus, G. thermoglucosidasius and G. thermodenitrificans. Int. J. Syst. Evol. Microbiol. 2001, 51, 433–446.
- Uemori, T.; Ishino, Y.; Fujita, K.; Asada, K.; Kato, I. Cloning of the DNA Polymerase Gene of Bacillus caldotenax and Characterization of the Gene Product. J. Biochem. 1993, 113, 401–410.
- Sellmann, E.; Schröder, K.L.; Knoblich, I.M.; Westermann, P. Purification and Characterization of DNA Polymerases from Bacillus Species. J. Bacteriol. 1992, 174, 4350–4355.
- Hayashizaki, Y.; Itoh, M.; Benno, Y.; Lezhava, A. Novel DNA Polymerase. U.S. Patent Application No. 20100047862A1, 25 February 2010.
- Oscorbin, I.P.; Boyarskikh, U.A.; Filipenko, M.L. Large Fragment of DNA Polymerase I from Geobacillus sp 777: Cloning and Comparison with DNA Polymerases I in Practical Applications. Mol. Biotechnol. 2015, 57, 947–959.
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-Mediated Isothermal Amplification of DNA. Nucleic Acids Res. 2000, 28, e63.
- Oscorbin, I.P.; Belousova, E.A.; Boyarskikh, U.A.; Zakabunin, A.I.; Khrapov, E.A.; Filipenko, M.L. Derivatives of Bst-Like Gss-Polymerase with Improved Processivity and Inhibitor Tolerance. Nucleic Acids Res. 2017, 45, 9595–9610.
- Kelly, R.M.; Adams, M.W. Metabolism in Hyperthermophilic Microorganisms. Antonie Van Leeuwenhoek 1994, 66, 247–270.
- Stetter, K.O. A Brief History of the Discovery of Hyperthermophilic Life. Biochem. Soc. Trans. 2013, 41, 416–420.
- Gelfand, D.H.; Lawyer, F.C. DNA Encoding a Thermostable Nucleic Acid Polymerase Enzyme from Thermotoga maritima. U.S. Patent No. 5,374,553, 20 December 1994.
- Gelfand, D.H.; Lawyer, F.C.; Stoffel, S. Mutated Thermostable Nucleic Acid Polymerase Enzyme from Thermotoga maritima. U.S. Patent No. 5,420,029, 30 May 1995.
- Diaz, R.S.; Sabino, E.C. Accuracy of Replication in the Polymerase Chain Reaction. Comparison between Thermotoga maritima DNA Polymerase and Thermus aquaticus DNA Polymerase. Brazilian J. Med. Biol. Res. 1998, 31, 1239–1242.
- Slater, M.R.; Huang, F.; Hartnett, J.R.; Bolchakova, E.; Storts, D.R.; Otto, P.; Miller, K.M.; Novikov, A.; Velikodvorskaya, G.A. Thermophilic DNA Polymerases from Thermotoga neapolitana. U.S. Patent No. 6,077,664, 20 June 2000.
- Yang, S.W.; Astatke, M.; Potter, J.; Chatterjee, D.K. Mutant Thermotoga neapolitana DNA Polymerase I: Altered Catalytic Properties for Non-Templated Nucleotide Addition and Incorporation of Correct Nucleotides. Nucleic Acids Res. 2002, 30, 4314–4320.
- Davalieva, K.G.; Efremov, G.D. A New Thermostable DNA Polymerase Mixture for Efficient Amplification of Long DNA Fragments. Appl. Biochem. Microbiol. 2010, 46, 230–234.
- Chang, J.R.; Choi, J.J.; Kim, H.K.; Kwon, S.T. Purification and Properties of Aquifex aeolicus DNA Polymerase Expressed in Escherichia coli. FEMS Microbiol. Lett. 2001, 201, 73–77.
- Takagi, M.; Nishioka, M.; Kakihara, H.; Kitabayashi, M.; Inoue, H.; Kawakami, B.; Oka, M.; Imanaka, T. Characterization of DNA Polymerase from Pyrococcus sp. Strain KOD1 and Its Application to PCR. Appl. Environ. Microbiol. 1997, 63, 4504–4510.
- Lundberg, K.S.; Shoemaker, D.D.; Adams, M.W.; Short, J.M.; Sorge, J.A.; Mathur, E.J. High-Fidelity Amplification Using a Thermostable DNA Polymerase Isolated from Pyrococcus furiosus. Gene 1991, 108, 1–6.
- Mattila, P.; Korpela, J.; Tenkanen, T.; Pitkänen, K. Fidelity of DNA Synthesis by the Thermococcus litoralis DNA Polymerase—An Extremely Heat Stable Enzyme with Proofreading Activity. Nucleic Acids Res. 1991, 19, 4967–4973.
- Atomi, H.; Fukui, T.; Kanai, T.; Morikawa, M.; Imanaka, T. Description of Thermococcus kodakaraensis sp. nov., a Well Studied Hyperthermophilic Archaeon Previously Reported as Pyrococcus sp. KOD1. Archaea 2004, 1, 263–267.
- Nishioka, M.; Mizuguchi, H.; Fujiwara, S.; Komatsubara, S.; Kitabayashi, M.; Uemura, H.; Takagi, M.; Imanaka, T. Long and Accurate PCR with a Mixture of KOD DNA Polymerase and Its Exonuclease Deficient Mutant Enzyme. J. Biotechnol. 2001, 88, 141–149.
- Southworth, M.W.; Kong, H.; Kucera, R.B.; Ware, J.; Jannasch, H.W.; Perler, F.B. Cloning of Thermostable DNA Polymerases from Hyperthermophilic Marine Archaea with Emphasis on Thermococcus sp. 9 Degrees N-7 and Mutations Affecting 3′-5′ Exonuclease Activity. Proc. Natl. Acad. Sci. USA 1996, 93, 5281–5285.
- Hopfner, K.P.; Eichinger, A.; Engh, R.A.; Laue, F.; Ankenbauer, W.; Huber, R.; Angerer, B. Crystal Structure of a Thermostable Type B DNA Polymerase from Thermococcus gorgonarius. Proc. Natl. Acad. Sci. USA 1999, 96, 3600–3605.
- Cambon-Bonavita, M.A.; Schmitt, P.; Zieger, M.; Flaman, J.M.; Lesongeur, F.; Raguénès, G.; Bindel, D.; Frisch, N.; Lakkis, Z.; Dupret, D.; et al. Cloning, Expression, and Characterization of DNA Polymerase I from the Hyperthermophilic Archaea Thermococcus fumicolans. Extremophiles 2000, 4, 215–225.
- Kim, Y.J.; Lee, H.S.; Bae, S.S.; Jeon, J.H.; Lim, J.K.; Cho, Y.; Nam, K.H.; Kang, S.G.; Kim, S.J.; Kwon, S.T.; et al. Cloning, Purification, and Characterization of a New DNA Polymerase from a Hyperthermophilic Archaeon, Thermococcus sp. NA1. J. Microbiol. Biotechnol. 2007, 17, 1090–1097.
- Lee, J.I.; Kim, Y.J.; Bae, H.; Cho, S.S.; Lee, J.H.; Kwon, S.T. Biochemical Properties and PCR Performance of a Family B DNA Polymerase from Hyperthermophilic Euryarchaeon Thermococcus peptonophilus. Appl. Biochem. Biotechnol. 2010, 160, 1585–1599.
- Griffiths, K.; Nayak, S.; Park, K.; Mandelman, D.; Modrell, B.; Lee, J.; Ng, B.; Gibbs, M.D.; Bergquist, P.L. New High Fidelity Polymerases from Thermococcus species. Protein Expr. Purif. 2007, 52, 19–30.
- Cho, S.S.; Kim, K.P.; Lee, K.K.; Youn, M.H.; Kwon, S.T. Characterization and PCR Application of a New High-Fidelity DNA Polymerase from Thermococcus waiotapuensis. Enzym. Microb. Technol. 2012, 51, 334–341.
- Cline, J.; Braman, J.C.; Hogrefe, H.H. PCR Fidelity of pfu DNA Polymerase and Other Thermostable DNA Polymerases. Nucleic Acids Res. 1996, 24, 3546–3551.
- Slupphaug, G.; Alseth, I.; Eftedal, I.; Volden, G.; Krokan, H.E. Low Incorporation of dUMP by Some Thermostable DNA Polymerases May Limit Their Use in PCR Amplifications. Anal. Biochem. 1993, 211, 164–169.
- Jannasch, H.W.; Wirsen, C.O.; Molyneaux, S.J.; Langworthy, T.A. Comparative Physiological Studies on Hyperthermophilic Archaea Isolated from Deep-Sea Hot Vents with Emphasis on Pyrococcus Strain GB-D. Appl. Environ. Microbiol. 1992, 58, 3472–3481.
- Huang, H.; Keohavong, P. Fidelity and Predominant Mutations Produced by Deep Vent Wild-Type and Exonuclease-Deficient DNA Polymerases during in Vitro DNA Amplification. DNA Cell Biol. 1996, 15, 589–594.
- Gueguen, Y.; Rolland, J.L.; Lecompte, O.; Azam, P.; Le Romancer, G.; Flament, D.; Raffin, J.P.; Dietrich, J. Characterization of Two DNA Polymerases from the Hyperthermophilic Euryarchaeon Pyrococcus abyssi. Eur. J. Biochem. 2001, 268, 5961–5969.
- Dietrich, J.; Schmitt, P.; Zieger, M.; Preve, B.; Rolland, J.L.; Chaabihi, H.; Gueguen, Y. PCR Performance of the Highly Thermostable Proof-Reading B-Type DNA Polymerase from Pyrococcus abyssi. FEMS Microbiol. Lett. 2002, 217, 89–94.
- Dabrowski, S.; Kur, J. Cloning and Expression in Escherichia coli of the Recombinant His-Tagged DNA Polymerases from Pyrococcus furiosus and Pyrococcus woesei. Protein Expr. Purif. 1998, 14, 131–138.
- Ghasemi, A.; Salmanian, A.H.; Sadeghifard, N.; Salarian, A.A.; Gholi, M.K. Cloning, Expression and Purification of Pwo Polymerase from Pyrococcus woesei. Iran. J. Microbiol. 2011, 3, 118–122.
- Hidajat, R.; McNicol, P. Primer-Directed Mutagenesis of an Intact Plasmid by Using Pwo DNA Polymerase in Long Distance Inverse PCR. Biotechniques 1997, 22, 32–34.
- Chen, T.; Romesberg, F.E. Directed Polymerase Evolution. FEBS Lett. 2014, 588, 219–229.
- Sun, L.; Ma, X.; Zhang, B.; Qin, Y.; Ma, J.; Du, Y.; Chen, T. From Polymerase Engineering to Semi-Synthetic Life: Artificial Expansion of the Central Dogma. RSC Chem. Biol. 2022, 3, 1173–1197.
- Coulther, T.A.; Stern, H.R.; Beuning, P.J. Engineering Polymerases for New Functions. Trends Biotechnol. 2019, 37, 1091–1103.
- Romero, P.A.; Arnold, F.H. Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876.
- Nikoomanzar, A.; Chim, N.; Yik, E.J.; Chaput, J.C. Engineering Polymerases for Applications in Synthetic Biology. Q. Rev. Biophys. 2020, 53, e8.
- McCullum, E.O.; Williams, B.A.; Zhang, J.L.; Chaput, J.C. Random Mutagenesis by Error-Prone PCR. Methods Mol. Biol. 2010, 634, 103–109.
- Stemmer, W.P. Rapid Evolution of a Protein in Vitro by DNA Shuffling. Nature 1994, 370, 389–391.
- Stemmer, W.P. DNA Shuffling by Random Fragmentation and Reassembly: In Vitro Recombination for Molecular Evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 10747–10751.
- Zhao, H.; Giver, L.; Shao, Z.; Affholter, J.A.; Arnold, F.H. Molecular Evolution by Staggered Extension Process (StEP) In Vitro Recombination. Nat. Biotechnol. 1998, 16, 258–261.
- Ness, J.E.; Kim, S.; Gottman, A.; Pak, R.; Krebber, A.; Borchert, T.V.; Govindarajan, S.; Mundorff, E.C.; Minshull, J. Synthetic Shuffling Expands Functional Protein Diversity by Allowing Amino Acids to Recombine Independently. Nat. Biotechnol. 2002, 20, 1251–1255.
- Milligan, J.N.; Garry, D.J. Shuffle Optimizer: A Program to Optimize DNA Shuffling for Protein Engineering. Methods Mol. Biol. 2017, 1472, 35–45.
- Milligan, J.N.; Shroff, R.; Garry, D.J.; Ellington, A.D. Evolution of a Thermophilic Strand-Displacing Polymerase Using High-Temperature Isothermal Compartmentalized Self-Replication. Biochemistry 2018, 57, 4607–4619.
- Huang, P.S.; Boyken, S.E.; Baker, D. The Coming of Age of De Novo Protein Design. Nature 2016, 537, 320–327.
- Marcos, E.; Silva, D.A. Essentials of De Novo Protein Design: Methods and Applications. Wires Comput. Mol. Sci. 2018, 8, e1374.
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More than 1000 Mutations. J. Mol. Biol. 2002, 320, 369–387.
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein Structure Prediction Using Rosetta. Methods Enzymol. 2004, 383, 66–93.
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting Stability Changes upon Mutation from the Protein Sequence or Structure. Nucleic Acids Res. 2005, 33, W306–W310.
- Wijma, H.J.; Floor, R.J.; Jekel, P.A.; Baker, D.; Marrink, S.J.; Janssen, D.B. Computationally Designed Libraries for Rapid Enzyme Stabilization. Protein Engi. Des. Sel. 2014, 27, 49–58.
- Goldenzweig, A.; Goldsmith, M.; Hill, S.E.; Gertman, O.; Laurino, P.; Ashani, Y.; Dym, O.; Unger, T.; Albeck, S.; Prilusky, J.; et al. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 2016, 63, 337–346.
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified Rational Protein Engineering with Sequence-Based Deep Representation Learning. Nat. Methods 2019, 16, 1315–1322.
- Paik, I.; Ngo, P.H.T.; Shroff, R.; Diaz, D.J.; Maranhao, A.C.; Walker, D.J.F.; Bhadra, S.; Ellington, A.D. Improved Bst DNA Polymerase Variants Derived via a Machine Learning Approach. Biochemistry 2021.
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-Learning-Guided Directed Evolution for Protein Engineering. Nat. Methods 2019, 16, 687–694.
- O’Maille, P.E.; Bakhtina, M.; Tsai, M.D. Structure-Based Combinatorial Protein Engineering (SCOPE). J Mol Biol. 2002, 321, 677–691.
- Reetz, M.T.; Bocola, M.; Carballeira, J.D.; Zha, D.; Vogel, A. Expanding the Range of Substrate Acceptance of Enzymes: Combinatorial Active-Site Saturation Test. Angew. Chem. Int. Edit. 2005, 44, 4192–4196.
- Reetz, M.T.; Carballeira, J.D. Iterative Saturation Mutagenesis (ISM) for Rapid Directed Evolution of Functional Enzymes. Nat. Protoc. 2007, 2, 891–903.
- Wong, T.S.; Tee, K.L.; Hauer, B.; Schwaneberg, U. Sequence Saturation Mutagenesis (SeSaM): A Novel Method for Directed Evolution. Nucleic Acids Res. 2004, 32, e26.
- Fox, R.J.; Davis, S.C.; Mundorff, E.C.; Newman, L.M.; Gavrilovic, V.; Ma, S.K.; Chung, L.M.; Ching, C.; Tam, S.; Muley, S.; et al. Improving Catalytic Function by ProSAR-Driven Enzyme Evolution. Nat. Biotechnol. 2007, 25, 338–344.
- Cole, M.F.; Gaucher, E.A. Exploiting Models of Molecular Evolution to Efficiently Direct Protein Engineering. J. Mol. Evol. 2011, 72, 193–203.
- Chen, T.; Hongdilokkul, N.; Liu, Z.; Adhikary, R.; Tsuen, S.S.; Romesberg, F.E. Evolution of Thermophilic DNA Polymerases for the Recognition and Amplification of C2′-Modified DNA. Nat. Chem. 2016, 8, 556–562.
- Markel, U.; Essani, K.D.; Besirlioglu, V.; Schiffels, J.; Streit, W.R.; Schwaneberg, U. Advances in Ultrahigh-Throughput Screening for Directed Enzyme Evolution. Chem. Soc. Rev. 2020, 49, 233–262.
- Esvelt, K.M.; Carlson, J.C.; Liu, D.R. A System for the Continuous Directed Evolution of Biomolecules. Nature 2011, 472, 499–503.
- Tawfik, D.S.; Griffiths, A.D. Man-Made Cell-Like Compartments for Molecular Evolution. Nat. Biotechnol. 1998, 16, 652–656.
- Ghadessy, F.J.; Ong, J.L.; Holliger, P. Directed Evolution of Polymerase Function by Compartmentalized Self-Replication. Proc. Natl. Acad. Sci. USA 2001, 98, 4552–4557.
- Ong, J.L.; Loakes, D.; Jaroslawski, S.; Too, K.; Holliger, P. Directed Evolution of DNA Polymerase, RNA Polymerase and Reverse Transcriptase Activity in a Single Polypeptide. J. Mol. Biol. 2006, 361, 537–550.
- Ellefson, J.W.; Gollihar, J.; Shroff, R.; Shivram, H.; Iyer, V.R.; Ellington, A.D. Synthetic Evolutionary Origin of a Proofreading Reverse Transcriptase. Science 2016, 352, 1590–1593.
- Shroff, R.; Ellefson, J.W.; Wang, S.S.; Boulgakov, A.A.; Hughes, R.A.; Ellington, A.D. Recovery of Information Stored in Modified DNA with an Evolved Polymerase. ACS Synth. Biol. 2022, 11, 554–561.
- Ellefson, J.W.; Meyer, A.J.; Hughes, R.A.; Cannon, J.R.; Brodbelt, J.S.; Ellington, A.D. Directed Evolution of Genetic Parts and Circuits by Compartmentalized Partnered Replication. Nat. Biotechnol. 2014, 32, 97–101.
- Pinheiro, V.B.; Taylor, A.I.; Cozens, C.; Abramov, M.; Renders, M.; Zhang, S.; Chaput, J.C.; Wengel, J.; Peak-Chew, S.Y.; McLaughlin, S.H.; et al. Synthetic Genetic Polymers Capable of Heredity and Evolution. Science 2012, 336, 341–344.
- Houlihan, G.; Arangundy-Franklin, S.; Porebski, B.T.; Subramanian, N.; Taylor, A.I.; Holliger, P. Discovery and Evolution of RNA and XNA Reverse Transcriptase Function and Fidelity. Nat. Chem. 2020, 12, 683–690.
- Paegel, B.M.; Joyce, G.F. Microfluidic Compartmentalized Directed Evolution. Chem. Biol. 2010, 17, 717–724.
- Larsen, A.C.; Dunn, M.R.; Hatch, A.; Sau, S.P.; Youngbull, C.; Chaput, J.C. A General Strategy for Expanding Polymerase Function by Droplet Microfluidics. Nat. Commun. 2016, 7, 11235.
- Price, A.K.; Paegel, B.M. Discovery in Droplets. Anal. Chem. 2016, 88, 339–353.
- Nikoomanzar, A.; Vallejo, D.; Chaput, J.C. Elucidating the Determinants of Polymerase Specificity by Microfluidic-Based Deep Mutational Scanning. ACS Synth. Biol. 2019, 8, 1421–1429.
- Taylor, A.I.; Houlihan, G.; Holliger, P. Beyond DNA and RNA: The Expanding Toolbox of Synthetic Genetics. Cold Spring Harbor Perspect. Biol. 2019, 11, a032490.
- Ohashi, S.; Hashiya, F.; Abe, H. Variety of Nucleotide Polymerase Mutants Aiming to Synthesize Modified RNA. ChemBioChem 2021, 22, 2398–2406.
- Sawai, H.; Ozaki-Nakamura, A.; Mine, M.; Ozaki, H. Synthesis of New Modified DNAs by Hyperthermophilic DNA Polymerase: Substrate and Template Specificity of Functionalized Thymidine Analogues Bearing an sp3-Hybridized Carbon at the C5 Alpha-Position for Several DNA Polymerases. Bioconjugate Chem. 2002, 13, 309–316.
- Kuwahara, M.; Nagashima, J.; Hasegawa, M.; Tamura, T.; Kitagata, R.; Hanawa, K.; Hososhima, S.; Kasamatsu, T.; Ozaki, H.; Sawai, H. Systematic Characterization of 2′-Deoxynucleoside-5′-triphosphate Analogs as Substrates for DNA Polymerases by Polymerase Chain Reaction and Kinetic Studies on Enzymatic Production of Modified DNA. Nucleic Acids Res. 2006, 34, 5383–5394.
- Mehedi Masud, M.; Ozaki-Nakamura, A.; Kuwahara, M.; Ozaki, H.; Sawai, H. Modified DNA Bearing 5(Methoxycarbonylmethyl)-2′-deoxyuridine: Preparation by PCR with Thermophilic DNA Polymerase and Postsynthetic Derivatization. ChemBioChem 2003, 4, 584–588.
- Hottin, A.; Marx, A. Structural Insights into the Processing of Nucleobase-Modified Nucleotides by DNA Polymerases. Accounts Chem. Res. 2016, 49, 418–427.
- Jager, S.; Rasched, G.; Kornreich-Leshem, H.; Engeser, M.; Thum, O.; Famulok, M. A Versatile Toolbox for Variable DNA Functionalization at High Density. J. Am. Chem. Soc. 2005, 127, 15071–15082.
- Baccaro, A.; Steck, A.L.; Marx, A. Barcoded Nucleotides. Angew. Chem. Int. Edit. 2012, 51, 254–257.
- Pochet, S.; Kaminski, P.A.; Van Aerschot, A.; Herdewijn, P.; Marlière, P. Replication of Hexitol Oligonucleotides as a Prelude to the Propagation of a Third Type of Nucleic Acid in Vivo. C. R. Biol. 2003, 326, 1175–1184.
- Jackson, L.N.; Chim, N.; Shi, C.H.; Chaput, J.C. Crystal Structures of a Natural DNA Polymerase that Functions as an XNA Reverse Transcriptase. Nucleic Acids Res. 2019, 47, 6973–6983.
- Tsai, C.H.; Chen, J.; Szostak, J.W. Enzymatic Synthesis of DNA on Glycerol Nucleic Acid Templates without Stable Duplex Formation between Product and Template. Proc. Natl. Acad. Sci. USA 2007, 104, 14598–14603.
- Kempeneers, V.; Renders, M.; Froeyen, M.; Herdewijn, P. Investigation of the DNA-Dependent Cyclohexenyl Nucleic Acid Polymerization and the Cyclohexenyl Nucleic Acid-Dependent DNA Polymerization. Nucleic Acids Res. 2005, 33, 3828–3836.
- Chaput, J.C.; Ichida, J.K.; Szostak, J.W. DNA Polymerase-Mediated DNA Synthesis on a TNA Template. J. Am. Chem. Soc. 2003, 125, 856–857.
- Peng, C.G.; Damha, M.J. Polymerase-Directed Synthesis of 2′-Deoxy-2′-fluoro-beta-D-arabinonucleic Acids. J. Am. Chem. Soc. 2007, 129, 5310–5311.
- Joyce, C.M. Choosing the Right Sugar: How Polymerases Select a Nucleotide Substrate. Proc. Natl. Acad. Sci. USA 1997, 94, 1619–1622.
- Anosova, I.; Kowai, E.A.; Dunn, M.R.; Chaput, J.C.; Van Horn, W.D.; Egli, M. The Structural Diversity of Artificial Genetic Polymers. Nucleic Acids Res. 2016, 44, 1007–1021.
- Gardner, A.F.; Jackson, K.M.; Boyle, M.M.; Buss, J.A.; Potapov, V.; Gehring, A.M.; Zatopek, K.M.; Corrêa, I.R., Jr.; Ong, J.L.; Jack, W.E. Therminator DNA Polymerase: Modified Nucleotides and Unnatural Substrates. Front. Mol. Biosci. 2019, 6, 28.
- Staiger, N.; Marx, A. A DNA Polymerase with Increased Reactivity for Ribonucleotides and C5-Modified Deoxyribonucleotides. ChemBioChem 2010, 11, 1963–1966.
- Ichida, J.K.; Horhota, A.; Zou, K.; McLaughlin, L.W.; Szostak, J.W. High Fidelity TNA Synthesis by Therminator Polymerase. Nucleic Acids Res. 2005, 33, 5219–5225.
- Fa, M.; Radeghieri, A.; Henry, A.A.; Romesberg, F.E. Expanding the Substrate Repertoire of a DNA Polymerase by Directed Evolution. J. Am. Chem. Soc. 2004, 126, 1748–1754.
- Chen, T.; Romesberg, F.E. Polymerase Chain Transcription: Exponential Synthesis of RNA and Modified RNA. J. Am. Chem. Soc. 2017, 139, 9949–9954.
- Song, P.; Zhang, R.; He, C.; Chen, T. Transcription, Reverse Transcription, and Amplification of Backbone-Modified Nucleic Acids with Laboratory-Evolved Thermophilic DNA Polymerases. Curr. Protoc. 2021, 1, e188.
- Chen, T.; Romesberg, F.E. Enzymatic Synthesis, Amplification, and Application of DNA with a Functionalized Backbone. Angew. Chem. Int. Edit. 2017, 56, 14046–14051.
- Arangundy-Franklin, S.; Taylor, A.I.; Porebski, B.T.; Genna, V.; Peak-Chew, S.; Vaisman, A.; Woodgate, R.; Orozco, M.; Holliger, P. A Synthetic Genetic Polymer with an Uncharged Backbone Chemistry Based on Alkyl Phosphonate Nucleic Acids. Nat. Chem. 2019, 11, 533–542.
- Dunn, M.R.; Otto, C.; Fenton, K.E.; Chaput, J.C. Improving Polymerase Activity with Unnatural Substrates by Sampling Mutations in Homologous Protein Architectures. ACS Chem. Biol. 2016, 11, 1210–1219.
- Nikoomanzar, A.; Vallejo, D.; Yik, E.J.; Chaput, J.C. Programmed Allelic Mutagenesis of a DNA Polymerase with Single Amino Acid Resolution. ACS Synth. Biol. 2020, 9, 1873–1881.
- Liu, C.; Cozens, C.; Jaziri, F.; Rozenski, J.; Marechal, A.; Dumbre, S.; Pezo, V.; Marlière, P.; Pinheiro, V.B.; Groaz, E.; et al. Phosphonomethyl Oligonucleotides as Backbone-Modified Artificial Genetic Polymers. J. Am. Chem. Soc. 2018, 140, 6690–6699.
- Hoshino, H.; Kasahara, Y.; Kuwahara, M.; Obika, S. DNA Polymerase Variants with High Processivity and Accuracy for Encoding and Decoding Locked Nucleic Acid Sequences. J. Am. Chem. Soc. 2020, 142, 21530–21537.
- Medina, E.; Yik, E.J.; Herdewijn, P.; Chaput, J.C. Functional Comparison of Laboratory-Evolved XNA Polymerases for Synthetic Biology. ACS Synth. Biol. 2021, 10, 1429–1437.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Revisions:
2 times
(View History)
Update Date:
09 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No