Nucleic acid polymerases are enzymes that catalyze DNA or RNA synthesis, including DNA polymerases (DNAPs), RNA polymerases (RNAPs), reverse transcriptases (RTs), and RNA-dependent RNA polymerases (RdRps), which play central roles in the storage and transmission of genetic information in living organisms, and have been widely applied in molecular biology and biotechnology. Their unique activities and functions have laid the foundation of many broadly used or modern techniques, including polymerase chain reaction (PCR), DNA sequencing, and DNA information storage. Thermostability is a desired property of nucleic acid polymerases for many of their applications, especially those involving thermocycling.
- thermophilic organisms
- thermophilic enzymes
- nucleic acid polymerases
1. Thermophilic and Hyperthermophilic Nucleic Acid Polymerases
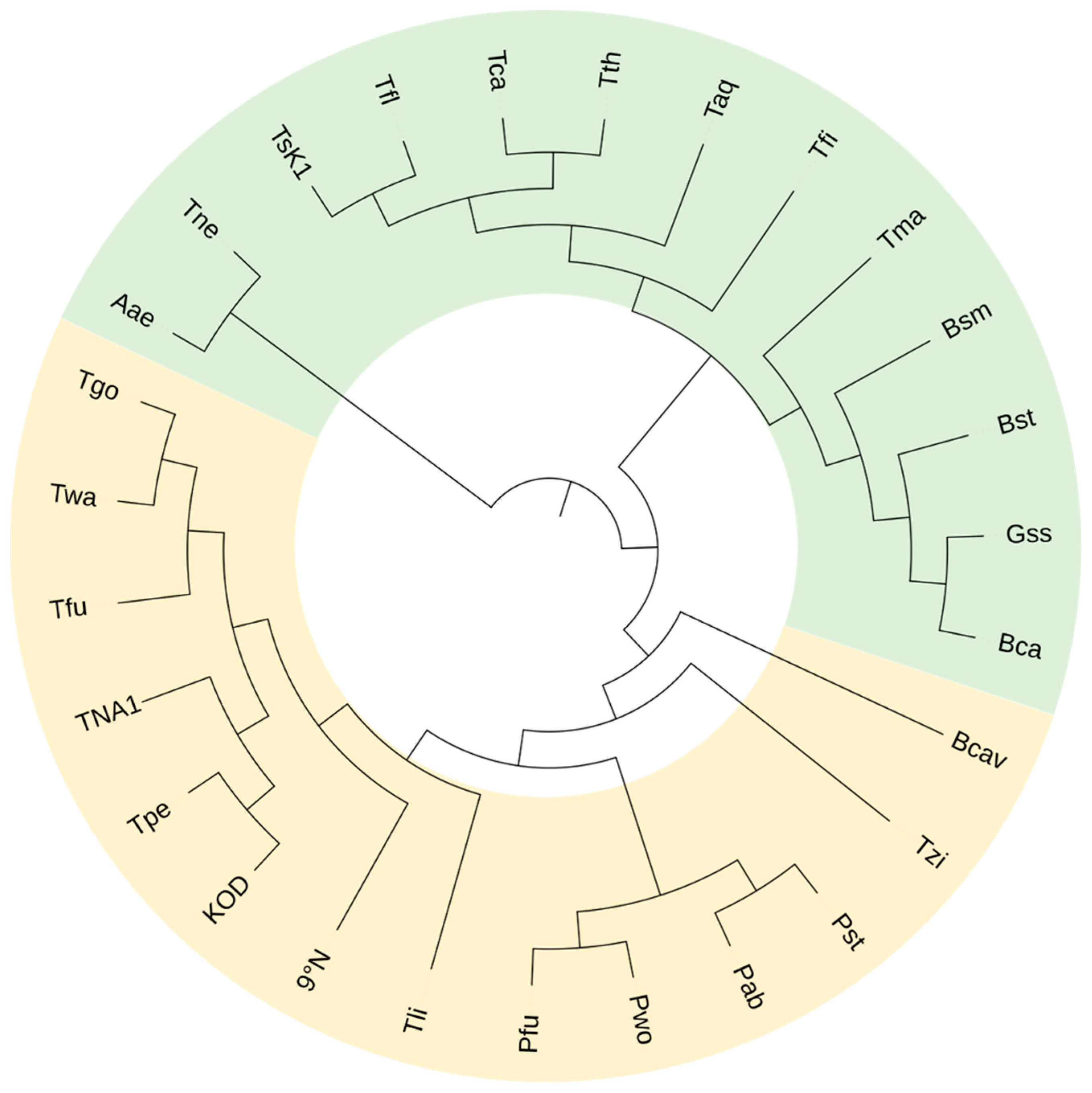
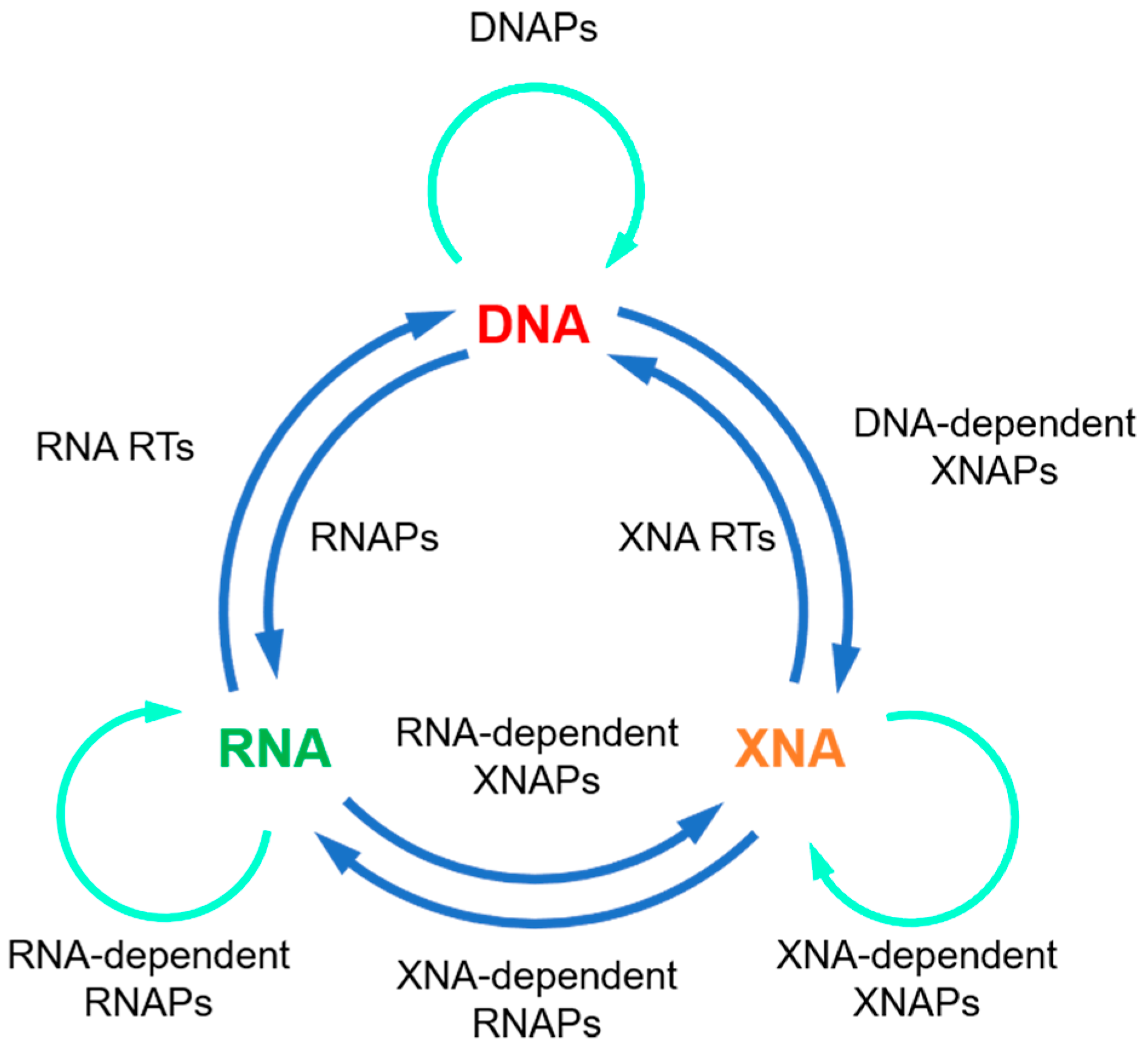
2. Strategies for Engineering Thermophilic Nucleic Acid Polymerases
2.1. Strategies for Mutant Generation or Library Construction
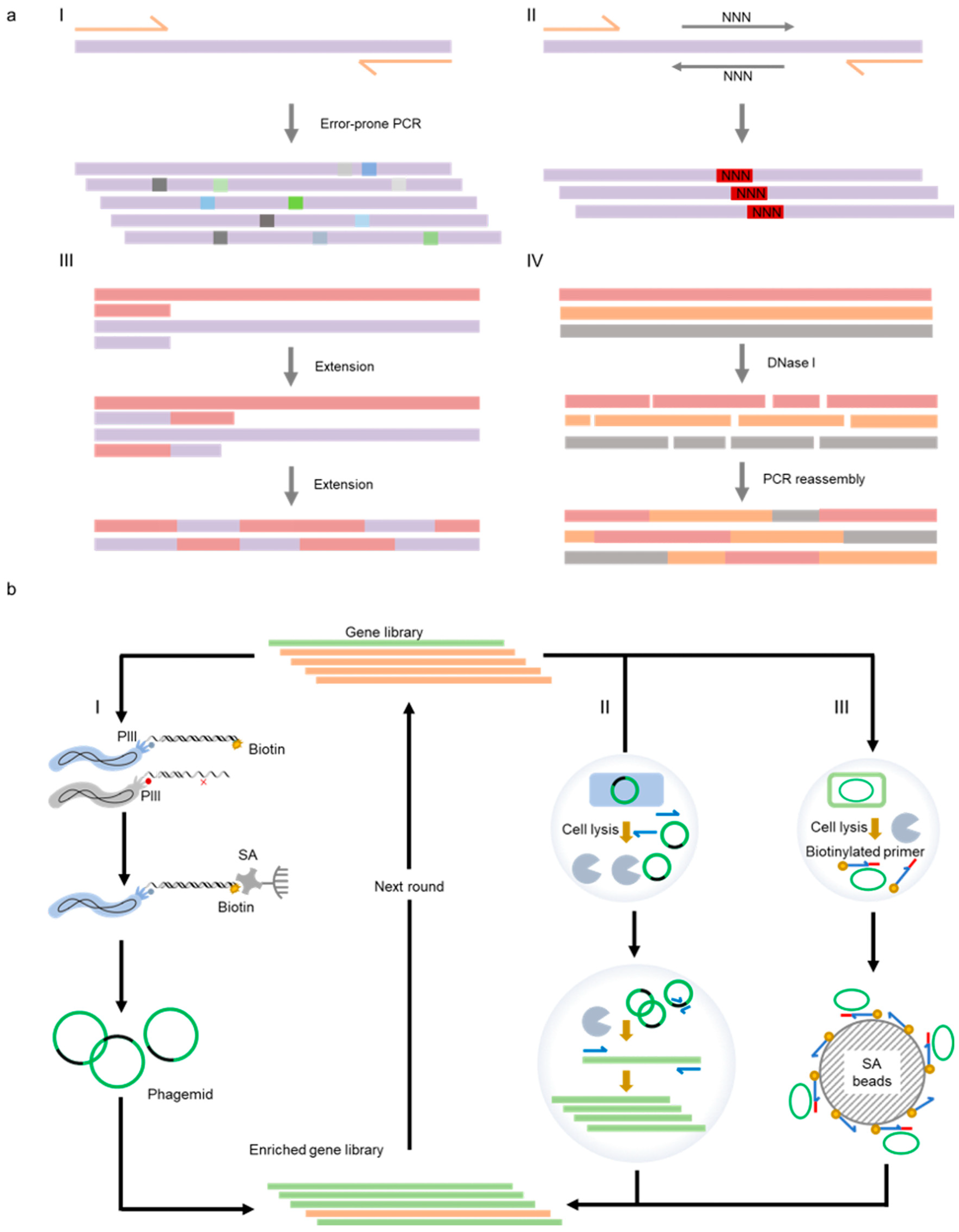
2.2. Strategies for the Selection or Screening of Polymerase Libraries
3. Thermophilic XNAPs
This entry is adapted from the peer-reviewed paper 10.3390/ijms232314969
References
- Laos, R.; Thomson, J.M.; Benner, S.A. DNA Polymerases Engineered by Directed Evolution to Incorporate Non-Standard Nucleotides. Front. Microbiol. 2014, 5, 565.
- Terpe, K. Overview of Thermostable DNA Polymerases for Classical PCR Applications: From Molecular and Biochemical Fundamentals to Commercial Systems. Appl. Microbiol. Biotechnol. 2013, 97, 10243–10254.
- Date, T.; Suzuki, K.; Imahori, K. Purification and Some Properties of DNA-Dependent RNA Polymerase from an Extreme Thermophile, Thermus thermophilus HB8. J. Biochem. 1975, 78, 845–858.
- Chien, A.; Edgar, D.B.; Trela, J.M. Deoxyribonucleic Acid Polymerase from the Extreme Thermophile Thermus aquaticus. J. Bacteriol. 1976, 127, 1550–1557.
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific Enzymatic Amplification of DNA In Vitro: The Polymerase Chain Reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–273.
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-Directed Enzymatic Amplification of DNA with a Thermostable DNA Polymerase. Science 1988, 239, 487–491.
- Lawyer, F.C.; Stoffel, S.; Saiki, R.K.; Chang, S.Y.; Landre, P.A.; Abramson, R.D.; Gelfand, D.H. High-Level Expression, Purification, and Enzymatic Characterization of Full-Length Thermus aquaticus DNA Polymerase and a Truncated form Deficient in 5′ to 3′ Exonuclease Activity. PCR Methods Appl. 1993, 2, 275–287.
- Tindall, K.R.; Kunkel, T.A. Fidelity of DNA Synthesis by the Thermus aquaticus DNA Polymerase. Biochemistry 1988, 27, 6008–6013.
- Eckert, K.A.; Kunkel, T.A. High Fidelity DNA Synthesis by the Thermus aquaticus DNA Polymerase. Nucleic Acids Res. 1990, 18, 3739–3744.
- Arezi, B.; Xing, W.; Sorge, J.A.; Hogrefe, H.H. Amplification Efficiency of Thermostable DNA Polymerases. Anal. Biochem. 2003, 321, 226–235.
- Barnes, W.M. The Fidelity of Taq Polymerase Catalyzing PCR Is Improved by an N-Terminal Deletion. Gene 1992, 112, 29–35.
- Grabko, V.I.; Chistyakova, L.G.; Lyapustin, V.N.; Korobko, V.G.; Miroshnikov, A.I. Reverse Transcription, Amplification and Sequencing of Poliovirus RNA by Taq DNA Polymerase. FEBS Lett. 1996, 387, 189–192.
- Bhadra, S.; Maranhao, A.C.; Paik, I.; Ellington, A.D. One-Enzyme Reverse Transcription qPCR Using Taq DNA Polymerase. Biochemistry 2020, 59, 4638–4645.
- Jung, S.E.; Choi, J.J.; Kim, H.K.; Kwon, S.T. Cloning and Analysis of the DNA Polymerase-Encoding Gene from Thermus filiformis. Mol. Cells. 1997, 7, 769–776.
- Dabrowski, S.; Kur, J. Recombinant His-Tagged DNA Polymerase. I. Cloning, Purification and Partial Characterization of Thermus thermophilus Recombinant DNA Polymerase. Acta Biochim. Pol. 1998, 45, 653–660.
- Akhmetzjanov, A.A.; Vakhitov, V.A. Molecular Cloning and Nucleotide Sequence of the DNA Polymerase Gene from Thermus flavus. Nucleic Acids Res. 1992, 20, 5839.
- Park, J.H.; Kim, J.S.; Kwon, S.T.; Lee, D.S. Purification and Characterization of Thermus caldophilus GK24 DNA Polymerase. Eur. J. Biochem. 1993, 214, 135–140.
- Saghatelyan, A.; Panosyan, H.; Trchounian, A.; Birkeland, N.K. Characteristics of DNA Polymerase I from an Extreme Thermophile, Thermus scotoductus Strain K1. MicrobiologyOpen 2021, 10, e1149.
- Choi, J.J.; Jung, S.E.; Kim, H.K.; Kwon, S.T. Purification and Properties of Thermus filiformis DNA Polymerase Expressed in Escherichia coli. Biotechnol. Appl. Biochem. 1999, 30, 19–25.
- Choi, J.J.; Kim, H.K.; Kwon, S.T. Purification and Characterization of the 5′→3′ Exonuclease Domain-Deleted Thermus filiformis DNA Polymerase Expressed in Escherichia coli. Biotechnol. Lett. 2001, 23, 1647–1652.
- Zheng, W.; Lee, J.E.; Potter, R.J.; Mandelman, D. DNA Polymerase Blends and Mutant DNA Polymerases. U.S. Patent Application No 11/170,762, 28 December 2006.
- Aye, S.L.; Fujiwara, K.; Ueki, A.; Doi, N. Engineering of DNA Polymerase I from Thermus thermophilus Using Compartmentalized Self-Replication. Biochem. Biophys. Res. Commun. 2018, 499, 170–176.
- Myers, T.W.; Gelfand, D.H. Reverse Transcription and DNA Amplification by a Thermus thermophilus DNA Polymerase. Biochemistry 1991, 30, 7661–7666.
- Choli, T.; Henning, P.; Wittmann-Liebold, B.; Reinhardt, R. Isolation, Characterization and Microsequence Analysis of a Small Basic Methylated DNA-Binding Protein from the Archaebacterium, Sulfolobus solfataricus. Biochim. Biophys. Acta. 1988, 950, 193–203.
- Sakai, H.D.; Kurosawa, N. Saccharolobus caldissimus Gen. nov., sp nov., a Facultatively Anaerobic Iron-Reducing Hyperthermophilic Archaeon Isolated from an Acidic Terrestrial Hot Spring, and Reclassification of Sulfolobus solfataricus as Saccharolobus solfataricus comb. Nov and Sulfolobus shibatae as Saccharolobus shibatae comb. nov. Int. J. Syst. Evol. Micr. 2018, 68, 1271–1278.
- Gao, Y.G.; Su, S.Y.; Robinson, H.; Padmanabhan, S.; Lim, L.; McCrary, B.S.; Edmondson, S.P.; Shriver, J.W.; Wang, A.H. The Crystal Structure of the Hyperthermophile Chromosomal Protein Sso7d Bound to DNA. Nat. Struct. Biol. 1998, 5, 782–786.
- Wang, Y.; Prosen, D.E.; Mei, L.; Sullivan, J.C.; Finney, M.; Vander Horn, P.B. A Novel Strategy to Engineer DNA Polymerases for Enhanced Processivity and Improved Performance in Vitro. Nucleic Acids Res. 2004, 32, 1197–1207.
- Harrell, R.A.; Hart, R.P. Rapid Preparation of Thermus flavus DNA Polymerase. PCR Methods Appl. 1994, 3, 372–375.
- Al-Soud, W.A.; Radstrom, P. Purification and Characterization of PCR-Inhibitory Components in Blood Cells. J. Clin. Microbiol. 2001, 39, 485–493.
- Wiedbrauk, D.L.; Werner, J.C.; Drevon, A.M. Inhibition of PCR by Aqueous and Vitreous Fluids. J. Clin. Microbiol. 1995, 33, 2643–2646.
- Kwon, S.T.; Kim, J.S.; Park, J.H.; Kim, H.K.; Lee, D.S. Cloning and Analysis of the DNA Polymerase-Encoding Gene from Thermus caldophilus GK24. Mol. Cells. 1997, 7, 264–271.
- Perler, F.B.; Kumar, S.; Kong, H. Thermostable DNA Polymerases. Adv. Protein Chem. 1996, 48, 377–435.
- Aliotta, J.M.; Pelletier, J.J.; Ware, J.L.; Moran, L.S.; Benner, J.S.; Kong, H. Thermostable Bst DNA Polymerase I Lacks a 3′→5′ Proofreading Exonuclease Activity. Genet. Anal. Biomol. Eng. 1996, 12, 185–195.
- Nazina, T.N.; Tourova, T.P.; Poltaraus, A.B.; Novikova, E.V.; Grigoryan, A.A.; Ivanova, A.E.; Lysenko, A.M.; Petrunyaka, V.V.; Osipov, G.A.; Belyaev, S.S.; et al. Taxonomic Study of Aerobic Thermophilic Bacilli: Descriptions of Geobacillus subterraneus Gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from Petroleum Reservoirs and Transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermodenitrificans to Geobacillus as the New Combinations G. stearothermophilus, G. thermocatenulatus, G. thermoleovorans, G. kaustophilus, G. thermoglucosidasius and G. thermodenitrificans. Int. J. Syst. Evol. Microbiol. 2001, 51, 433–446.
- Uemori, T.; Ishino, Y.; Fujita, K.; Asada, K.; Kato, I. Cloning of the DNA Polymerase Gene of Bacillus caldotenax and Characterization of the Gene Product. J. Biochem. 1993, 113, 401–410.
- Sellmann, E.; Schröder, K.L.; Knoblich, I.M.; Westermann, P. Purification and Characterization of DNA Polymerases from Bacillus Species. J. Bacteriol. 1992, 174, 4350–4355.
- Hayashizaki, Y.; Itoh, M.; Benno, Y.; Lezhava, A. Novel DNA Polymerase. U.S. Patent Application No. 20100047862A1, 25 February 2010.
- Oscorbin, I.P.; Boyarskikh, U.A.; Filipenko, M.L. Large Fragment of DNA Polymerase I from Geobacillus sp 777: Cloning and Comparison with DNA Polymerases I in Practical Applications. Mol. Biotechnol. 2015, 57, 947–959.
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-Mediated Isothermal Amplification of DNA. Nucleic Acids Res. 2000, 28, e63.
- Oscorbin, I.P.; Belousova, E.A.; Boyarskikh, U.A.; Zakabunin, A.I.; Khrapov, E.A.; Filipenko, M.L. Derivatives of Bst-Like Gss-Polymerase with Improved Processivity and Inhibitor Tolerance. Nucleic Acids Res. 2017, 45, 9595–9610.
- Kelly, R.M.; Adams, M.W. Metabolism in Hyperthermophilic Microorganisms. Antonie Van Leeuwenhoek 1994, 66, 247–270.
- Stetter, K.O. A Brief History of the Discovery of Hyperthermophilic Life. Biochem. Soc. Trans. 2013, 41, 416–420.
- Gelfand, D.H.; Lawyer, F.C. DNA Encoding a Thermostable Nucleic Acid Polymerase Enzyme from Thermotoga maritima. U.S. Patent No. 5,374,553, 20 December 1994.
- Gelfand, D.H.; Lawyer, F.C.; Stoffel, S. Mutated Thermostable Nucleic Acid Polymerase Enzyme from Thermotoga maritima. U.S. Patent No. 5,420,029, 30 May 1995.
- Diaz, R.S.; Sabino, E.C. Accuracy of Replication in the Polymerase Chain Reaction. Comparison between Thermotoga maritima DNA Polymerase and Thermus aquaticus DNA Polymerase. Brazilian J. Med. Biol. Res. 1998, 31, 1239–1242.
- Slater, M.R.; Huang, F.; Hartnett, J.R.; Bolchakova, E.; Storts, D.R.; Otto, P.; Miller, K.M.; Novikov, A.; Velikodvorskaya, G.A. Thermophilic DNA Polymerases from Thermotoga neapolitana. U.S. Patent No. 6,077,664, 20 June 2000.
- Yang, S.W.; Astatke, M.; Potter, J.; Chatterjee, D.K. Mutant Thermotoga neapolitana DNA Polymerase I: Altered Catalytic Properties for Non-Templated Nucleotide Addition and Incorporation of Correct Nucleotides. Nucleic Acids Res. 2002, 30, 4314–4320.
- Davalieva, K.G.; Efremov, G.D. A New Thermostable DNA Polymerase Mixture for Efficient Amplification of Long DNA Fragments. Appl. Biochem. Microbiol. 2010, 46, 230–234.
- Chang, J.R.; Choi, J.J.; Kim, H.K.; Kwon, S.T. Purification and Properties of Aquifex aeolicus DNA Polymerase Expressed in Escherichia coli. FEMS Microbiol. Lett. 2001, 201, 73–77.
- Takagi, M.; Nishioka, M.; Kakihara, H.; Kitabayashi, M.; Inoue, H.; Kawakami, B.; Oka, M.; Imanaka, T. Characterization of DNA Polymerase from Pyrococcus sp. Strain KOD1 and Its Application to PCR. Appl. Environ. Microbiol. 1997, 63, 4504–4510.
- Lundberg, K.S.; Shoemaker, D.D.; Adams, M.W.; Short, J.M.; Sorge, J.A.; Mathur, E.J. High-Fidelity Amplification Using a Thermostable DNA Polymerase Isolated from Pyrococcus furiosus. Gene 1991, 108, 1–6.
- Mattila, P.; Korpela, J.; Tenkanen, T.; Pitkänen, K. Fidelity of DNA Synthesis by the Thermococcus litoralis DNA Polymerase—An Extremely Heat Stable Enzyme with Proofreading Activity. Nucleic Acids Res. 1991, 19, 4967–4973.
- Atomi, H.; Fukui, T.; Kanai, T.; Morikawa, M.; Imanaka, T. Description of Thermococcus kodakaraensis sp. nov., a Well Studied Hyperthermophilic Archaeon Previously Reported as Pyrococcus sp. KOD1. Archaea 2004, 1, 263–267.
- Nishioka, M.; Mizuguchi, H.; Fujiwara, S.; Komatsubara, S.; Kitabayashi, M.; Uemura, H.; Takagi, M.; Imanaka, T. Long and Accurate PCR with a Mixture of KOD DNA Polymerase and Its Exonuclease Deficient Mutant Enzyme. J. Biotechnol. 2001, 88, 141–149.
- Southworth, M.W.; Kong, H.; Kucera, R.B.; Ware, J.; Jannasch, H.W.; Perler, F.B. Cloning of Thermostable DNA Polymerases from Hyperthermophilic Marine Archaea with Emphasis on Thermococcus sp. 9 Degrees N-7 and Mutations Affecting 3′-5′ Exonuclease Activity. Proc. Natl. Acad. Sci. USA 1996, 93, 5281–5285.
- Hopfner, K.P.; Eichinger, A.; Engh, R.A.; Laue, F.; Ankenbauer, W.; Huber, R.; Angerer, B. Crystal Structure of a Thermostable Type B DNA Polymerase from Thermococcus gorgonarius. Proc. Natl. Acad. Sci. USA 1999, 96, 3600–3605.
- Cambon-Bonavita, M.A.; Schmitt, P.; Zieger, M.; Flaman, J.M.; Lesongeur, F.; Raguénès, G.; Bindel, D.; Frisch, N.; Lakkis, Z.; Dupret, D.; et al. Cloning, Expression, and Characterization of DNA Polymerase I from the Hyperthermophilic Archaea Thermococcus fumicolans. Extremophiles 2000, 4, 215–225.
- Kim, Y.J.; Lee, H.S.; Bae, S.S.; Jeon, J.H.; Lim, J.K.; Cho, Y.; Nam, K.H.; Kang, S.G.; Kim, S.J.; Kwon, S.T.; et al. Cloning, Purification, and Characterization of a New DNA Polymerase from a Hyperthermophilic Archaeon, Thermococcus sp. NA1. J. Microbiol. Biotechnol. 2007, 17, 1090–1097.
- Lee, J.I.; Kim, Y.J.; Bae, H.; Cho, S.S.; Lee, J.H.; Kwon, S.T. Biochemical Properties and PCR Performance of a Family B DNA Polymerase from Hyperthermophilic Euryarchaeon Thermococcus peptonophilus. Appl. Biochem. Biotechnol. 2010, 160, 1585–1599.
- Griffiths, K.; Nayak, S.; Park, K.; Mandelman, D.; Modrell, B.; Lee, J.; Ng, B.; Gibbs, M.D.; Bergquist, P.L. New High Fidelity Polymerases from Thermococcus species. Protein Expr. Purif. 2007, 52, 19–30.
- Cho, S.S.; Kim, K.P.; Lee, K.K.; Youn, M.H.; Kwon, S.T. Characterization and PCR Application of a New High-Fidelity DNA Polymerase from Thermococcus waiotapuensis. Enzym. Microb. Technol. 2012, 51, 334–341.
- Cline, J.; Braman, J.C.; Hogrefe, H.H. PCR Fidelity of pfu DNA Polymerase and Other Thermostable DNA Polymerases. Nucleic Acids Res. 1996, 24, 3546–3551.
- Slupphaug, G.; Alseth, I.; Eftedal, I.; Volden, G.; Krokan, H.E. Low Incorporation of dUMP by Some Thermostable DNA Polymerases May Limit Their Use in PCR Amplifications. Anal. Biochem. 1993, 211, 164–169.
- Jannasch, H.W.; Wirsen, C.O.; Molyneaux, S.J.; Langworthy, T.A. Comparative Physiological Studies on Hyperthermophilic Archaea Isolated from Deep-Sea Hot Vents with Emphasis on Pyrococcus Strain GB-D. Appl. Environ. Microbiol. 1992, 58, 3472–3481.
- Huang, H.; Keohavong, P. Fidelity and Predominant Mutations Produced by Deep Vent Wild-Type and Exonuclease-Deficient DNA Polymerases during in Vitro DNA Amplification. DNA Cell Biol. 1996, 15, 589–594.
- Gueguen, Y.; Rolland, J.L.; Lecompte, O.; Azam, P.; Le Romancer, G.; Flament, D.; Raffin, J.P.; Dietrich, J. Characterization of Two DNA Polymerases from the Hyperthermophilic Euryarchaeon Pyrococcus abyssi. Eur. J. Biochem. 2001, 268, 5961–5969.
- Dietrich, J.; Schmitt, P.; Zieger, M.; Preve, B.; Rolland, J.L.; Chaabihi, H.; Gueguen, Y. PCR Performance of the Highly Thermostable Proof-Reading B-Type DNA Polymerase from Pyrococcus abyssi. FEMS Microbiol. Lett. 2002, 217, 89–94.
- Dabrowski, S.; Kur, J. Cloning and Expression in Escherichia coli of the Recombinant His-Tagged DNA Polymerases from Pyrococcus furiosus and Pyrococcus woesei. Protein Expr. Purif. 1998, 14, 131–138.
- Ghasemi, A.; Salmanian, A.H.; Sadeghifard, N.; Salarian, A.A.; Gholi, M.K. Cloning, Expression and Purification of Pwo Polymerase from Pyrococcus woesei. Iran. J. Microbiol. 2011, 3, 118–122.
- Hidajat, R.; McNicol, P. Primer-Directed Mutagenesis of an Intact Plasmid by Using Pwo DNA Polymerase in Long Distance Inverse PCR. Biotechniques 1997, 22, 32–34.
- Chen, T.; Romesberg, F.E. Directed Polymerase Evolution. FEBS Lett. 2014, 588, 219–229.
- Sun, L.; Ma, X.; Zhang, B.; Qin, Y.; Ma, J.; Du, Y.; Chen, T. From Polymerase Engineering to Semi-Synthetic Life: Artificial Expansion of the Central Dogma. RSC Chem. Biol. 2022, 3, 1173–1197.
- Coulther, T.A.; Stern, H.R.; Beuning, P.J. Engineering Polymerases for New Functions. Trends Biotechnol. 2019, 37, 1091–1103.
- Romero, P.A.; Arnold, F.H. Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876.
- Nikoomanzar, A.; Chim, N.; Yik, E.J.; Chaput, J.C. Engineering Polymerases for Applications in Synthetic Biology. Q. Rev. Biophys. 2020, 53, e8.
- McCullum, E.O.; Williams, B.A.; Zhang, J.L.; Chaput, J.C. Random Mutagenesis by Error-Prone PCR. Methods Mol. Biol. 2010, 634, 103–109.
- Stemmer, W.P. Rapid Evolution of a Protein in Vitro by DNA Shuffling. Nature 1994, 370, 389–391.
- Stemmer, W.P. DNA Shuffling by Random Fragmentation and Reassembly: In Vitro Recombination for Molecular Evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 10747–10751.
- Zhao, H.; Giver, L.; Shao, Z.; Affholter, J.A.; Arnold, F.H. Molecular Evolution by Staggered Extension Process (StEP) In Vitro Recombination. Nat. Biotechnol. 1998, 16, 258–261.
- Ness, J.E.; Kim, S.; Gottman, A.; Pak, R.; Krebber, A.; Borchert, T.V.; Govindarajan, S.; Mundorff, E.C.; Minshull, J. Synthetic Shuffling Expands Functional Protein Diversity by Allowing Amino Acids to Recombine Independently. Nat. Biotechnol. 2002, 20, 1251–1255.
- Milligan, J.N.; Garry, D.J. Shuffle Optimizer: A Program to Optimize DNA Shuffling for Protein Engineering. Methods Mol. Biol. 2017, 1472, 35–45.
- Milligan, J.N.; Shroff, R.; Garry, D.J.; Ellington, A.D. Evolution of a Thermophilic Strand-Displacing Polymerase Using High-Temperature Isothermal Compartmentalized Self-Replication. Biochemistry 2018, 57, 4607–4619.
- Huang, P.S.; Boyken, S.E.; Baker, D. The Coming of Age of De Novo Protein Design. Nature 2016, 537, 320–327.
- Marcos, E.; Silva, D.A. Essentials of De Novo Protein Design: Methods and Applications. Wires Comput. Mol. Sci. 2018, 8, e1374.
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More than 1000 Mutations. J. Mol. Biol. 2002, 320, 369–387.
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein Structure Prediction Using Rosetta. Methods Enzymol. 2004, 383, 66–93.
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting Stability Changes upon Mutation from the Protein Sequence or Structure. Nucleic Acids Res. 2005, 33, W306–W310.
- Wijma, H.J.; Floor, R.J.; Jekel, P.A.; Baker, D.; Marrink, S.J.; Janssen, D.B. Computationally Designed Libraries for Rapid Enzyme Stabilization. Protein Engi. Des. Sel. 2014, 27, 49–58.
- Goldenzweig, A.; Goldsmith, M.; Hill, S.E.; Gertman, O.; Laurino, P.; Ashani, Y.; Dym, O.; Unger, T.; Albeck, S.; Prilusky, J.; et al. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 2016, 63, 337–346.
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified Rational Protein Engineering with Sequence-Based Deep Representation Learning. Nat. Methods 2019, 16, 1315–1322.
- Paik, I.; Ngo, P.H.T.; Shroff, R.; Diaz, D.J.; Maranhao, A.C.; Walker, D.J.F.; Bhadra, S.; Ellington, A.D. Improved Bst DNA Polymerase Variants Derived via a Machine Learning Approach. Biochemistry 2021.
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-Learning-Guided Directed Evolution for Protein Engineering. Nat. Methods 2019, 16, 687–694.
- O’Maille, P.E.; Bakhtina, M.; Tsai, M.D. Structure-Based Combinatorial Protein Engineering (SCOPE). J Mol Biol. 2002, 321, 677–691.
- Reetz, M.T.; Bocola, M.; Carballeira, J.D.; Zha, D.; Vogel, A. Expanding the Range of Substrate Acceptance of Enzymes: Combinatorial Active-Site Saturation Test. Angew. Chem. Int. Edit. 2005, 44, 4192–4196.
- Reetz, M.T.; Carballeira, J.D. Iterative Saturation Mutagenesis (ISM) for Rapid Directed Evolution of Functional Enzymes. Nat. Protoc. 2007, 2, 891–903.
- Wong, T.S.; Tee, K.L.; Hauer, B.; Schwaneberg, U. Sequence Saturation Mutagenesis (SeSaM): A Novel Method for Directed Evolution. Nucleic Acids Res. 2004, 32, e26.
- Fox, R.J.; Davis, S.C.; Mundorff, E.C.; Newman, L.M.; Gavrilovic, V.; Ma, S.K.; Chung, L.M.; Ching, C.; Tam, S.; Muley, S.; et al. Improving Catalytic Function by ProSAR-Driven Enzyme Evolution. Nat. Biotechnol. 2007, 25, 338–344.
- Cole, M.F.; Gaucher, E.A. Exploiting Models of Molecular Evolution to Efficiently Direct Protein Engineering. J. Mol. Evol. 2011, 72, 193–203.
- Chen, T.; Hongdilokkul, N.; Liu, Z.; Adhikary, R.; Tsuen, S.S.; Romesberg, F.E. Evolution of Thermophilic DNA Polymerases for the Recognition and Amplification of C2′-Modified DNA. Nat. Chem. 2016, 8, 556–562.
- Markel, U.; Essani, K.D.; Besirlioglu, V.; Schiffels, J.; Streit, W.R.; Schwaneberg, U. Advances in Ultrahigh-Throughput Screening for Directed Enzyme Evolution. Chem. Soc. Rev. 2020, 49, 233–262.
- Esvelt, K.M.; Carlson, J.C.; Liu, D.R. A System for the Continuous Directed Evolution of Biomolecules. Nature 2011, 472, 499–503.
- Tawfik, D.S.; Griffiths, A.D. Man-Made Cell-Like Compartments for Molecular Evolution. Nat. Biotechnol. 1998, 16, 652–656.
- Ghadessy, F.J.; Ong, J.L.; Holliger, P. Directed Evolution of Polymerase Function by Compartmentalized Self-Replication. Proc. Natl. Acad. Sci. USA 2001, 98, 4552–4557.
- Ong, J.L.; Loakes, D.; Jaroslawski, S.; Too, K.; Holliger, P. Directed Evolution of DNA Polymerase, RNA Polymerase and Reverse Transcriptase Activity in a Single Polypeptide. J. Mol. Biol. 2006, 361, 537–550.
- Ellefson, J.W.; Gollihar, J.; Shroff, R.; Shivram, H.; Iyer, V.R.; Ellington, A.D. Synthetic Evolutionary Origin of a Proofreading Reverse Transcriptase. Science 2016, 352, 1590–1593.
- Shroff, R.; Ellefson, J.W.; Wang, S.S.; Boulgakov, A.A.; Hughes, R.A.; Ellington, A.D. Recovery of Information Stored in Modified DNA with an Evolved Polymerase. ACS Synth. Biol. 2022, 11, 554–561.
- Ellefson, J.W.; Meyer, A.J.; Hughes, R.A.; Cannon, J.R.; Brodbelt, J.S.; Ellington, A.D. Directed Evolution of Genetic Parts and Circuits by Compartmentalized Partnered Replication. Nat. Biotechnol. 2014, 32, 97–101.
- Pinheiro, V.B.; Taylor, A.I.; Cozens, C.; Abramov, M.; Renders, M.; Zhang, S.; Chaput, J.C.; Wengel, J.; Peak-Chew, S.Y.; McLaughlin, S.H.; et al. Synthetic Genetic Polymers Capable of Heredity and Evolution. Science 2012, 336, 341–344.
- Houlihan, G.; Arangundy-Franklin, S.; Porebski, B.T.; Subramanian, N.; Taylor, A.I.; Holliger, P. Discovery and Evolution of RNA and XNA Reverse Transcriptase Function and Fidelity. Nat. Chem. 2020, 12, 683–690.
- Paegel, B.M.; Joyce, G.F. Microfluidic Compartmentalized Directed Evolution. Chem. Biol. 2010, 17, 717–724.
- Larsen, A.C.; Dunn, M.R.; Hatch, A.; Sau, S.P.; Youngbull, C.; Chaput, J.C. A General Strategy for Expanding Polymerase Function by Droplet Microfluidics. Nat. Commun. 2016, 7, 11235.
- Price, A.K.; Paegel, B.M. Discovery in Droplets. Anal. Chem. 2016, 88, 339–353.
- Nikoomanzar, A.; Vallejo, D.; Chaput, J.C. Elucidating the Determinants of Polymerase Specificity by Microfluidic-Based Deep Mutational Scanning. ACS Synth. Biol. 2019, 8, 1421–1429.
- Taylor, A.I.; Houlihan, G.; Holliger, P. Beyond DNA and RNA: The Expanding Toolbox of Synthetic Genetics. Cold Spring Harbor Perspect. Biol. 2019, 11, a032490.
- Ohashi, S.; Hashiya, F.; Abe, H. Variety of Nucleotide Polymerase Mutants Aiming to Synthesize Modified RNA. ChemBioChem 2021, 22, 2398–2406.
- Sawai, H.; Ozaki-Nakamura, A.; Mine, M.; Ozaki, H. Synthesis of New Modified DNAs by Hyperthermophilic DNA Polymerase: Substrate and Template Specificity of Functionalized Thymidine Analogues Bearing an sp3-Hybridized Carbon at the C5 Alpha-Position for Several DNA Polymerases. Bioconjugate Chem. 2002, 13, 309–316.
- Kuwahara, M.; Nagashima, J.; Hasegawa, M.; Tamura, T.; Kitagata, R.; Hanawa, K.; Hososhima, S.; Kasamatsu, T.; Ozaki, H.; Sawai, H. Systematic Characterization of 2′-Deoxynucleoside-5′-triphosphate Analogs as Substrates for DNA Polymerases by Polymerase Chain Reaction and Kinetic Studies on Enzymatic Production of Modified DNA. Nucleic Acids Res. 2006, 34, 5383–5394.
- Mehedi Masud, M.; Ozaki-Nakamura, A.; Kuwahara, M.; Ozaki, H.; Sawai, H. Modified DNA Bearing 5(Methoxycarbonylmethyl)-2′-deoxyuridine: Preparation by PCR with Thermophilic DNA Polymerase and Postsynthetic Derivatization. ChemBioChem 2003, 4, 584–588.
- Hottin, A.; Marx, A. Structural Insights into the Processing of Nucleobase-Modified Nucleotides by DNA Polymerases. Accounts Chem. Res. 2016, 49, 418–427.
- Jager, S.; Rasched, G.; Kornreich-Leshem, H.; Engeser, M.; Thum, O.; Famulok, M. A Versatile Toolbox for Variable DNA Functionalization at High Density. J. Am. Chem. Soc. 2005, 127, 15071–15082.
- Baccaro, A.; Steck, A.L.; Marx, A. Barcoded Nucleotides. Angew. Chem. Int. Edit. 2012, 51, 254–257.
- Pochet, S.; Kaminski, P.A.; Van Aerschot, A.; Herdewijn, P.; Marlière, P. Replication of Hexitol Oligonucleotides as a Prelude to the Propagation of a Third Type of Nucleic Acid in Vivo. C. R. Biol. 2003, 326, 1175–1184.
- Jackson, L.N.; Chim, N.; Shi, C.H.; Chaput, J.C. Crystal Structures of a Natural DNA Polymerase that Functions as an XNA Reverse Transcriptase. Nucleic Acids Res. 2019, 47, 6973–6983.
- Tsai, C.H.; Chen, J.; Szostak, J.W. Enzymatic Synthesis of DNA on Glycerol Nucleic Acid Templates without Stable Duplex Formation between Product and Template. Proc. Natl. Acad. Sci. USA 2007, 104, 14598–14603.
- Kempeneers, V.; Renders, M.; Froeyen, M.; Herdewijn, P. Investigation of the DNA-Dependent Cyclohexenyl Nucleic Acid Polymerization and the Cyclohexenyl Nucleic Acid-Dependent DNA Polymerization. Nucleic Acids Res. 2005, 33, 3828–3836.
- Chaput, J.C.; Ichida, J.K.; Szostak, J.W. DNA Polymerase-Mediated DNA Synthesis on a TNA Template. J. Am. Chem. Soc. 2003, 125, 856–857.
- Peng, C.G.; Damha, M.J. Polymerase-Directed Synthesis of 2′-Deoxy-2′-fluoro-beta-D-arabinonucleic Acids. J. Am. Chem. Soc. 2007, 129, 5310–5311.
- Joyce, C.M. Choosing the Right Sugar: How Polymerases Select a Nucleotide Substrate. Proc. Natl. Acad. Sci. USA 1997, 94, 1619–1622.
- Anosova, I.; Kowai, E.A.; Dunn, M.R.; Chaput, J.C.; Van Horn, W.D.; Egli, M. The Structural Diversity of Artificial Genetic Polymers. Nucleic Acids Res. 2016, 44, 1007–1021.
- Gardner, A.F.; Jackson, K.M.; Boyle, M.M.; Buss, J.A.; Potapov, V.; Gehring, A.M.; Zatopek, K.M.; Corrêa, I.R., Jr.; Ong, J.L.; Jack, W.E. Therminator DNA Polymerase: Modified Nucleotides and Unnatural Substrates. Front. Mol. Biosci. 2019, 6, 28.
- Staiger, N.; Marx, A. A DNA Polymerase with Increased Reactivity for Ribonucleotides and C5-Modified Deoxyribonucleotides. ChemBioChem 2010, 11, 1963–1966.
- Ichida, J.K.; Horhota, A.; Zou, K.; McLaughlin, L.W.; Szostak, J.W. High Fidelity TNA Synthesis by Therminator Polymerase. Nucleic Acids Res. 2005, 33, 5219–5225.
- Fa, M.; Radeghieri, A.; Henry, A.A.; Romesberg, F.E. Expanding the Substrate Repertoire of a DNA Polymerase by Directed Evolution. J. Am. Chem. Soc. 2004, 126, 1748–1754.
- Chen, T.; Romesberg, F.E. Polymerase Chain Transcription: Exponential Synthesis of RNA and Modified RNA. J. Am. Chem. Soc. 2017, 139, 9949–9954.
- Song, P.; Zhang, R.; He, C.; Chen, T. Transcription, Reverse Transcription, and Amplification of Backbone-Modified Nucleic Acids with Laboratory-Evolved Thermophilic DNA Polymerases. Curr. Protoc. 2021, 1, e188.
- Chen, T.; Romesberg, F.E. Enzymatic Synthesis, Amplification, and Application of DNA with a Functionalized Backbone. Angew. Chem. Int. Edit. 2017, 56, 14046–14051.
- Arangundy-Franklin, S.; Taylor, A.I.; Porebski, B.T.; Genna, V.; Peak-Chew, S.; Vaisman, A.; Woodgate, R.; Orozco, M.; Holliger, P. A Synthetic Genetic Polymer with an Uncharged Backbone Chemistry Based on Alkyl Phosphonate Nucleic Acids. Nat. Chem. 2019, 11, 533–542.
- Dunn, M.R.; Otto, C.; Fenton, K.E.; Chaput, J.C. Improving Polymerase Activity with Unnatural Substrates by Sampling Mutations in Homologous Protein Architectures. ACS Chem. Biol. 2016, 11, 1210–1219.
- Nikoomanzar, A.; Vallejo, D.; Yik, E.J.; Chaput, J.C. Programmed Allelic Mutagenesis of a DNA Polymerase with Single Amino Acid Resolution. ACS Synth. Biol. 2020, 9, 1873–1881.
- Liu, C.; Cozens, C.; Jaziri, F.; Rozenski, J.; Marechal, A.; Dumbre, S.; Pezo, V.; Marlière, P.; Pinheiro, V.B.; Groaz, E.; et al. Phosphonomethyl Oligonucleotides as Backbone-Modified Artificial Genetic Polymers. J. Am. Chem. Soc. 2018, 140, 6690–6699.
- Hoshino, H.; Kasahara, Y.; Kuwahara, M.; Obika, S. DNA Polymerase Variants with High Processivity and Accuracy for Encoding and Decoding Locked Nucleic Acid Sequences. J. Am. Chem. Soc. 2020, 142, 21530–21537.
- Medina, E.; Yik, E.J.; Herdewijn, P.; Chaput, J.C. Functional Comparison of Laboratory-Evolved XNA Polymerases for Synthetic Biology. ACS Synth. Biol. 2021, 10, 1429–1437.