Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giuseppe Murdaca | -- | 1905 | 2022-12-08 09:35:10 | | | |
| 2 | Sirius Huang | Meta information modification | 1905 | 2022-12-08 10:01:17 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Furci, F.; Murdaca, G.; Allegra, A.; Gammeri, L.; Senna, G.; Gangemi, S. Role of IL-33 Signalling in COVID-19 Inflammatory Status. Encyclopedia. Available online: https://encyclopedia.pub/entry/38287 (accessed on 26 June 2026).
Furci F, Murdaca G, Allegra A, Gammeri L, Senna G, Gangemi S. Role of IL-33 Signalling in COVID-19 Inflammatory Status. Encyclopedia. Available at: https://encyclopedia.pub/entry/38287. Accessed June 26, 2026.
Furci, Fabiana, Giuseppe Murdaca, Alessandro Allegra, Luca Gammeri, Gianenrico Senna, Sebastiano Gangemi. "Role of IL-33 Signalling in COVID-19 Inflammatory Status" Encyclopedia, https://encyclopedia.pub/entry/38287 (accessed June 26, 2026).
Furci, F., Murdaca, G., Allegra, A., Gammeri, L., Senna, G., & Gangemi, S. (2022, December 08). Role of IL-33 Signalling in COVID-19 Inflammatory Status. In Encyclopedia. https://encyclopedia.pub/entry/38287
Furci, Fabiana, et al. "Role of IL-33 Signalling in COVID-19 Inflammatory Status." Encyclopedia. Web. 08 December, 2022.
Copy Citation
During acute infection, the increased levels of pro-inflammatory cytokines, which are involved in the pathology of disease and the development of SARS-CoV-2-induced acute respiratory disease syndrome, the life-threatening form of this infection, are correlated with patient survival and disease severity. IL-33, a key cytokine involved in both innate and adaptive immune responses in mucosal organs, can increase airway inflammation, mucus secretion and Th2 cytokine synthesis in the lungs, following respiratory infections. Similar to cases of exposure to known respiratory virus infections, exposure to SARS-CoV-2 induces the expression of IL-33, correlating with T-cell activation and lung disease severity.
IL-33
COVID-19
cytokine storm
inflammation
1. Introduction
The involvement of various molecules, cytokines and immune cells has been described in COVID-19 (Figure 1), but a key concept to highlight is that the cells damaged by SARS-CoV-2 release alarmins, or damage-associated molecular patterns (DAMPs), which act as danger molecules in promoting inflammatory response [1][2]. IL-33, as reported above, is recognized as an alarmin, an expression of cellular damage or infection, whose increased levels in epithelial and endothelial cells recall its pro-inflammatory role in respiratory diseases. In particular, the mature bioactive form of IL-33 requires cleavage by proteases. In the literature, it has been reported that serum IL-33 levels are up-regulated in elderly patients affected by COVID-19, an expression of the epithelial damage induced by the interaction between the airway epithelium and activated immune cells, linking them to severe outcomes [3]. These data imply a key role for IL-33 in COVID-19 pathogenesis. However, SARS-CoV-2-derived papain-like protease (PLpro), an inducer of IL-33 in epithelial cells, could also trigger epithelium-derived IL-33, inducing inflammation in the lungs.
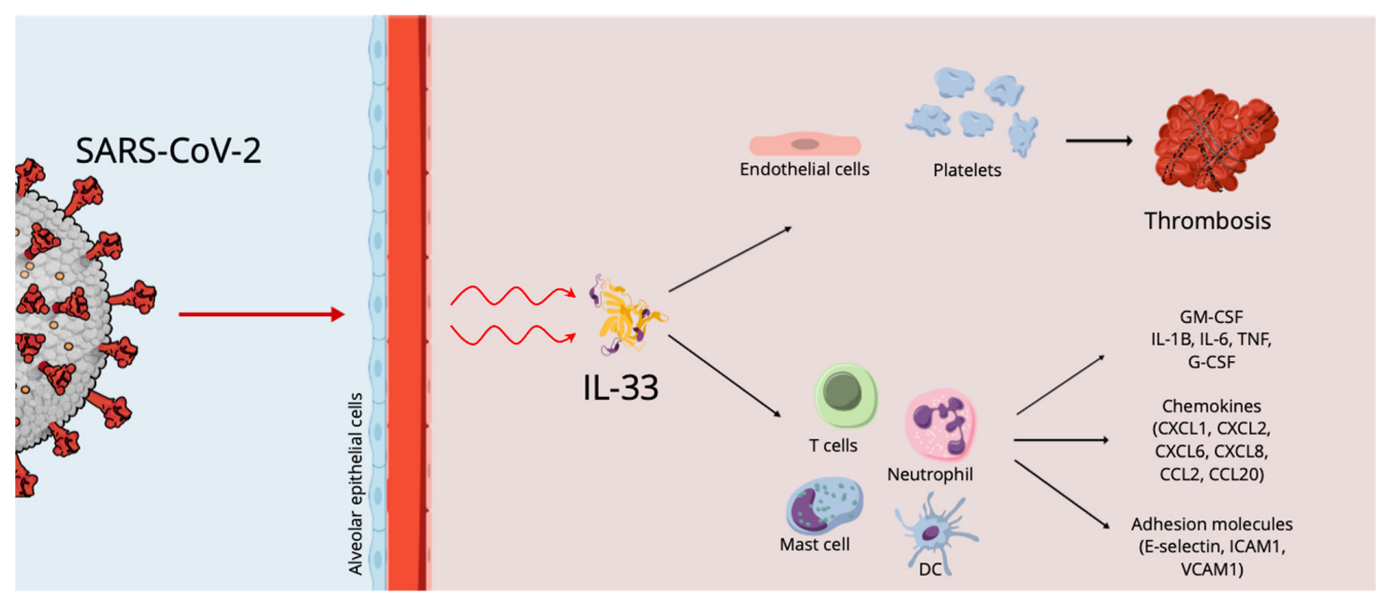
Figure 1. The effects of SARS-CoV-2 on the alveolar epithelium.
It is equally important to highlight that IL-33 can stimulate antiviral cytotoxic T-cell action and the production of antibodies. Stanczak MA. et al., further highlighting the role of IL-33 in COVID-19 immunobiology, reported that the persistent production of IL-33 in response to T-cell activation may be useful in the case of later contact with the virus [4].
Liang et al. infected two human epithelial cell lines, Fadu and LS513, with SARS-CoV-2 in vitro, reporting a significant increase in IL-33 transcript levels in both cell lines at 72 h post-infection [3].
Marcovic SS. et al. analysed the correlation of IL-33 and other innate immunity cytokines with COVID-19 severity in patients with COVID-19 that were divided into two groups (mild/moderate and severe/critical). The authors reported that in a more progressive stage of the disease, increased IL-33 levels are a determinant in lung inflammation favouring the production of innate pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α, IL-12 and IL-23 in several target cells, causing the most severe forms of COVID-19. The authors also reported a positive correlation between IL-33 and clinical parameters of COVID-19, in particular higher values of the neutrophil count, C-reactive protein (CRP), D dimer, procalcitonin (PCT), lactate dehydrogenase (LDH), urea, creatinine, creatine kinase CK, ferritin, total bilirubin (TBIL) and aspartate aminotransferase (AST), as well as lower values of lymphocyte and monocyte count and albumin. Serum levels > 332.08 pg/mL of IL-33 were considered a factor related to the risk of increased severity of COVID-19 [5]. The same results were reported by Wang et al. and by Rubio-Sánchez et al. [6][7].
Meidaninikjeh S. et al. and Miyazawa M. reported that cellular compositions of lung infiltrates in patients with COVID-19 pneumonia are different depending on the progression of the disease. In detail, in patients with moderate pneumonia, infiltrates are made up mainly of lymphoid and dendritic cells, while in patients with the severe form of the disease there is a massive infiltration of macrophages and neutrophils [8][9]. IL-33 induces neutrophil migration via macrophage-derived CXCL1 and CXCL2, whereas neutrophil elastase and cathepsin G are responsible for IL-33 processing and maturation, which leads to inflammatory responses. In agreement with these data, Marcovic SS. et al. highlighted an increased neutrophil count in patients with severe COVID-19 [5].
Focusing on seroconversion, in the literature, it has been reported that IL-33 clustered together with anti-receptor binding domain (RBD) IgG in moderate and severe cases [10]. Stanczak M.A. et al. reported that the SARS-CoV-2-infection-induced production of IL-33 clustered together with IgG titres and that after recovery from COVID-19 patients still had persistent circulating PBMCs that produced IL-33 in response to virus-specific T-cell activation, correlating with seropositivity [4]. Zeng G. et al. studied soluble ST2 (sST2, a cardiac biomarker) levels among COVID-19 patients and a possible relationship between inflammatory status and disease severity, highlighting a positive correlation between serum sST2 levels and CRP, and a negative correlation between CD4+ and CD8+ T lymphocyte counts and sST2. In particular, Zeng Z. et al. analysed the relationship of serum sST2 to lymphocyte subsets, reporting that CD3+CD4+ and CD3+CD8+ lymphocyte absolute counts were negatively correlated with the levels of serum sST2 in patients with COVID-19, highlighting that the elevated serum sST2 may favour the dysfunction of T cells in COVID-19 progression [11]. This concurs with many studies reporting that during COVID-19 infection patients are often characterized by low lymphocyte counts and impaired cytotoxic activity [12]. Gaurav et al. compared lung sections of COVID-19 patients to those of normal patients and patients with chronic inflammatory lung diseases, such as IPF and COPD, highlighting that tissue IL-33 levels were higher among the latter. Indeed, patients with COVID-19 had very low IL-33 expression, which was significantly reduced compared to that of control subjects. IL-33 levels were increased in post-COVID fibrosis and were higher compared to the levels in patients affected by COPD and IPF [13].
2. Cytokine Storm, IL-33 Effects and Thrombosis in COVID 19 Infection
The most frequent complication of systemic infections is the triggering of the coagulation process, which can exhibit a wide variety of clinical symptoms fluctuating from subclinical expression, represented by increased laboratory indicators for fibrin and thrombin products, to the appearance of thrombosis and disseminated intravascular coagulation [14].
Regarding COVID-19 patients, the devastating occurrence of uncontrolled coagulopathy is a powerful prognosticator of mortality in infected subjects [15]. However, the genesis of hypercoagulable condition and thrombosis correlated to COVID-19 is uncertain [16][17][18]. COVID-19 infection likely provokes a condition of immune system hyperstimulation, which has been defined as immunothrombosis (Figure 2), in which stimulated neutrophils react with activated platelets and the coagulation factors, causing intravascular clot generation in all vessels. It is supposed that the overstated immunothrombosis that occurs within pulmonary microvessels is the leading cause of COVID-19 symptoms [19][20].

Figure 2. Mechanisms responsible for immunothrombosis in SARS-CoV-2 infection. TF = tissue factor.
A different motive for stimulating the coagulation cascade is represented by endothelial cell damage and activation by SARS-CoV-2 infection itself. Assessment of skin and pulmonary autopsies of subjects who died of COVID-19 infection demonstrated the presence of thrombosis and microvascular damage, coherent with severe, widespread stimulation of alternative and lectin-founded paths of the complement system [21] and successive stimulation of the clotting cascade, provoking fibrin accumulation [22]. The resulting hypercoagulable condition is the main reason for venous thromboembolism, pulmonary embolism and deep venous thrombosis of the lower extremities, which has been reported in subjects affected by COVID-19.
Recently, an in vitro experiment confirmed the role of the cytokine storm in the generation of thrombosis in COVID-19 patients [23], and IL-33 plays an essential role in the cytokine storm, especially regarding the triggering of pro-thrombotic phenomena.
Indeed, IL-33 stimulates endothelial cells (ECs), causing the onset of an inflammatory phenotype via an increase in different molecules such as intercellular adhesion molecule-1, vascular cell adhesion molecule-1, monocyte chemoattractant protein-1 and endothelial-selectin or cytokines such as IL-6 and Il-8 [24][25]. IL-33 also supports angiogenesis and regulates the proteolytic ability of ECs by easing the generation of plasminogen activator inhibitor type-1 and urokinase-type plasminogen activator [26][27]. In a different patient setting, several reports showed that IL-33 serum concentrations are correlated with reduced survival in subjects with ST elevation myocardial infarction [28][29], while an experiment demonstrated that increased levels of IL-33 after coronary stent implantation are correlated with coronary restenosis [30].
Furthermore, tissue factor (TF) is the main factor of blood coagulation and has a critical effect in the onset of thrombotic events. IL-33 stimulated TF mRNA and protein in umbilical and coronary artery ECs in an ST2- and NF-κB-dependent modality, but this increase was IL-1-independent. IL-33-exposed ECs decreased the coagulation time of plasma and whole blood, while in atherosclerotic plaques, TF mRNA was related to IL-33 mRNA generation. Moreover, IL-33 and TF protein were found in the same location of clot generation in plaques of subjects with carotid stenosis. Thus, IL-33 could increase thrombotic ability and operate on thrombus formation via the production of TF in ECs [31].
However, the effects of IL-33 on TF production could also occur via other mechanisms. Monocytes and monocyte-derived microvesicles (MVs) are one of the principal origins of TF. Increased monocyte-derived TF and an increased circulating amount of procoagulant MVs participate in the establishment of a prothrombotic state. IL-33 was reported to induce a time- and dose-dependent increase in monocyte TF concentrations. IL-33-exposed monocytes also released CD14+TF+ MVs and IL-33 was reported to increase the TF action of monocytes and monocyte-originated MVs. Intermediate monocytes (IMs) displayed the greatest receptor expression, with a reduced expression on non-classical monocytes (NCMs) and classical monocytes (CMs). IL-33 caused a relevant increase in TF only in the IM, while a reduced expression was reported in NCMs, and no increase in CMs was noted. Thus, IL-33 may further participate in the establishment of a prothrombotic condition [32].
Beside the potent effect on TF generation, in vitro experiments demonstrated IL-33 was able to reduce the production of TF pathway inhibitor in ECs. This effect influences the coagulation time of whole blood and plasma ex vivo [33]. Furthermore, IL-33-caused generation of adhesion molecules on ECs could cause an increased binding and stimulation of white cells and leukocyte-originated MVs to ECs [34][35][36].
Furthermore, the strict, bidirectional relationships between IL-33 and platelets and their precursors are of particular significance. A study performed on an animal experimental model of intestinal inflammation displayed the precise effect of IL-33 in stimulating platelets. Inflammation increases systemic platelet triggering and blood coagulation, generating increased concentrations of IL-33 and provoking platelet activation through a rise in 5-HT release. More relevantly, authors assessed that deficiency of epithelial-originated IL-33 reduced 5-HT concentration, causing an altered platelet activation [37].
Finally, though platelets are the most relevant effectors of thrombosis, they are also immune cells. Platelets interact with immune cells, modifying their activity, maturation and proliferation, interfering with all expression of the immune response. Megakaryocytes (Mks) are the bone marrow precursors of circulating platelets, and until lately Mks were only believed to be bone-marrow-occupant cells. However, Mks also are present in the lung, and these Mks present higher numbers of immune molecules than bone marrow Mks. A study attempted to assess the immune activities of lung Mks (MkLs). They reported that MkLs had gene expression profiles that are analogous to antigen-presenting cells, and the immune phenotype was variable and regulated by the tissue immune milieu, as demonstrated by bone marrow Mks presenting an MkL-like phenotype after exposure to immune molecules, such as IL-33. In vitro and in vivo experiments showed that MkL assumed and managed antigenic proteins and pathogens, stimulated CD4+T cell triggering in an MHC II-dependent modality [38] and may have a central role in IL-33-induced thrombosis in COVID 19 patients.
A direct or indirect intervention aimed at modulating the effects of the IL-33 on the homeostasis of the coagulative system could guarantee an effective prophylactic or therapeutic action in the catastrophic thrombosis induced by the cytokine storm that complicates the course of COVID-19 infection.
References
- Allegra, A.; Di Gioacchino, M.; Tonacci, A.; Musolino, C. Immunopathology of SARS-CoV-2 Infection: Immune Cells and Mediators, Prognostic Factors, and Immune-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 4782.
- Di Salvo, E.; Di Gioacchino, M.; Tonacci, A.; Casciaro, M.; Gangemi, S. Alarmins, COVID-19 and comorbidities. Ann. Med. 2021, 53, 777–785.
- Liang, Y.; Ge, Y.; Sun, J. IL-33 in COVID-19: Friend or foe? Cell. Mol. Immunol. 2021, 18, 1602–1604.
- Stanczak, M.A.; Sanin, D.E.; Apostolova, P.; Nerz, G.; Lampaki, D.; Hofmann, M.; Steimann, D.; Krohn-Grimberghe, M.; Thimme, R.; Mittler, G.; et al. IL-33 expression in response to SARS-CoV-2 correlates with seropositivity in COVID-19 convalescent individuals. Nat. Commun. 2021, 12, 2133.
- Markovic, S.S.; Jovanovic, M.; Gajovic, N.; Jurisevic, M.; Arsenijevic, N.; Jovanovic, M.; Jovanovic, M.; Mijailovic, Z.; Lukic, S.; Zornic, N.; et al. IL 33 Correlates With COVID-19 Severity, Radiographic and Clinical Finding. Front. Med. 2021, 8, 749569.
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069.
- Rubio-Sánchez, R.; Lepe-Balsalobre, E.; Viloria-Peñas, M. Prognostic factors for the severity of SARS-CoV-2 infection. Adv. Lab. Med. 2021, 2, 253–258.
- Meidaninikjeh, S.; Sabouni, N.; Marzouni, H.Z.; Bengar, S.; Khalili, A.; Jafari, R. Monocytes and macrophages in COVID-19: Friends and foes. Life Sci. 2021, 269, 119010.
- Miyazawa, M. Immunopathogenesis of SARS-CoV-2-induced pneumonia:lessons from influenza virus infection. Inflamm. Regen. 2020, 40, 39.
- Murdaca, G.; Paladin, F.; Tonacci, A.; Borro, M.; Greco, M.; Gerosa, A.; Isola, S.; Allegra, A.; Gangemi, S. Involvement of Il-33 in the Pathogenesis and Prognosis of Major Respiratory Viral Infections: Future Perspectives for Personalized Therapy. Biomedicines 2022, 10, 715.
- Zeng, Z.; Hong, X.Y.; Li, Y.; Chen, W.; Ye, G.; Li, Y.; Luo, Y. Serum-soluble ST2 as a novel biomarker reflecting inflammatory status and illness severity in patients with COVID-19. Biomark. Med. 2020, 14, 1619–1629.
- Cristinziano, L.; Poto, R.; Criscuolo, G.; Ferrara, A.L.; Galdiero, M.R.; Modestino, L.; Loffredo, S.; de Paulis, A.; Marone, G.; Spadaro, G.; et al. IL-33 and Superantigenic Activation of Human Lung Mast Cells Induce the Release of Angiogenic and Lymphangiogenic Factors. Cells 2021, 10, 145.
- Gaurav, R.; Anderson, D.R.; Radio, S.J.; Bailey, K.L.; England, B.R.; Mikuls, T.R.; Thiele, G.M.; Strah, H.M.; Romberger, D.J.; Wyatt, T.A.; et al. IL-33 Depletion in COVID-19 Lungs. Chest 2021, 160, 1656–1659.
- Opal, S.M. Interactions between coagulation and inflammation. Scand. J. Infect. Dis. 2003, 35, 545–554.
- Wifi, M.N.; Morad, M.A.; El Sheemy, R.; Abdeen, N.; Afify, S.; Abdalgaber, M.; Abdellatef, A.; Zaghloul, M.; Alboraie, M.; El-Kassas, M. Hemostatic system and COVID-19 crosstalk: A review of the available evidence. World J. Methodol. 2022, 12, 331–349.
- Nappi, F.; Nappi, P.; Gambardella, I.; Avtaar Singh, S.S. Thromboembolic Disease and Cardiac Thrombotic Complication in COVID-19: A Systematic Review. Metabolites 2022, 12, 889.
- Allegra, A.; Innao, V.; Allegra, A.G.; Musolino, C. Coagulopathy and thromboembolic events in patients with SARS-CoV-2 infection: Pathogenesis and management strategies. Ann. Hematol. 2020, 99, 1953–1965.
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649.
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329.
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587.
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13.
- Chaturvedi, S.; Braunstein, E.M.; Yuan, X.; Yu, J.; Alexander, A.; Chen, H.; Gavriilaki, E.; Alluri, R.; Streiff, M.B.; Petri, M.; et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood 2020, 135, 239–251.
- Gurbel, P.A.; Tantry, U.S. In Vitro Evidence for the Role of Cytokine Storm in the Generation of Stent Thrombosis in COVID-19 Patients. Cardiovasc. Revasc. Med. 2022, 35, 139–140.
- Demyanets, S.; Konya, V.; Kastl, S.P.; Kaun, C.; Rauscher, S.; Niessner, A.; Pentz, R.; Pfaffenberger, S.; Rychli, K.; Lemberger, C.E.; et al. Interleukin-33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2080–2089.
- Aoki, S.; Hayakawa, M.; Ozaki, H.; Takezako, N.; Obata, H.; Ibaraki, N.; Tsuru, T.; Tominaga, S.; Yanagisawa, K. ST2 gene expression is proliferation-dependent and its ligand, IL-33, induces inflammatory reaction in endothelial cells. Mol. Cell. Biochem. 2010, 335, 75–81.
- Choi, Y.S.; Choi, H.J.; Min, J.K.; Pyun, B.J.; Maeng, Y.S.; Park, H.; Kim, J.; Kim, Y.M.; Kwon, Y.G. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood 2009, 114, 3117–3126.
- Stojkovic, S.; Kaun, C.; Heinz, M.; Krychtiuk, K.A.; Rauscher, S.; Lemberger, C.E.; de Martin, R.; Gröger, M.; Petzelbauer, P.; Huk, I.; et al. Interleukin-33 induces urokinase in human endothelial cells--possible impact on angiogenesis. J. Thromb. Haemost. 2014, 12, 948–957.
- Demyanets, S.; Speidl, W.S.; Tentzeris, I.; Jarai, R.; Katsaros, K.M.; Farhan, S.; Krychtiuk, K.A.; Wonnerth, A.; Weiss, T.W.; Huber, K.; et al. Soluble ST2 and interleukin-33 levels in coronary artery disease: Relation to disease activity and adverse outcome. PLoS ONE. 2014, 9, e95055.
- Dhillon, O.S.; Narayan, H.K.; Khan, S.Q.; Kelly, D.; Quinn, P.A.; Squire, I.B.; Davies, J.E.; Ng, L.L. Pre-discharge risk stratification in unselected STEMI: Is there a role for ST2 or its natural ligand IL-33 when compared with contemporary risk markers? Int. J. Cardiol. 2013, 167, 2182–2188.
- Demyanets, S.; Tentzeris, I.; Jarai, R.; Katsaros, K.M.; Farhan, S.; Wonnerth, A.; Weiss, T.W.; Wojta, J.; Speidl, W.S.; Huber, K. An increase of interleukin-33 serum levels after coronary stent implantation is associated with coronary in-stent restenosis. Cytokine 2014, 67, 65–70.
- Stojkovic, S.; Kaun, C.; Basilio, J.; Rauscher, S.; Hell, L.; Krychtiuk, K.A.; Bonstingl, C.; de Martin, R.; Gröger, M.; Ay, C.; et al. Tissue factor is induced by interleukin-33 in human endothelial cells: A new link between coagulation and inflammation. Sci. Rep. 2016, 6, 25171.
- Stojkovic, S.; Thulin, Å.; Hell, L.; Thaler, B.; Rauscher, S.; Baumgartner, J.; Gröger, M.; Ay, C.; Demyanets, S.; Neumayer, C.; et al. IL-33 stimulates the release of procoagulant microvesicles from human monocytes and differentially increases tissue factor in human monocyte subsets. Thromb. Haemost. 2017, 117, 1379–1390.
- Barton, L.M.; Duval, E.; Stroberg, E.; Gosh, S.; Mukhopadhyay, S. COVID-19 autopsies, Oklahoma, U.S.A. Am. J. Clin. Pathol. 2020, 153, 725–733.
- Zeyda, M.; Wernly, B.; Demyanets, S.; Kaun, C.; Hammerle, M.; Hantusch, B.; Schranz, M.; Neuhofer, A.; Itariu, B.K.; Keck, M.; et al. Severe obesity increases adipose tissue expression of interleukin-33 and its receptor ST2, both predominantly detectable in endothelial cells of human adipose tissue. Int. J. Obes. 2013, 37, 658–665.
- Van Hinsbergh, V.W. Endothelium-role in regulation of coagulation and inflammation. Semin. Immunopathol. 2012, 34, 93–106.
- Demyanets, S.; Stojkovic, S.; Huber, K.; Wojta, J. The Paradigm Change of IL-33 in Vascular Biology. Int. J. Mol. Sci. 2021, 22, 13288.
- Chen, Z.; Luo, J.; Li, J.; Kim, G.; Stewart, A.; Huang, Y.; Wu, C. Intestinal IL-33 promotes platelet activity for neutrophil recruitment during acute inflammation. Blood 2022, 139, 1878–1891.
- Pariser, D.N.; Hilt, Z.T.; Ture, S.K.; Blick-Nitko, S.K.; Looney, M.R.; Cleary, S.J.; Roman-Pagan, E.; Saunders, J., II; Georas, S.N.; Veazey, J.; et al. Lung megakaryocytes are immune modulatory cells. J. Clin. Investig. 2021, 131, e137377.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
878
Revisions:
2 times
(View History)
Update Date:
08 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No