During acute infection, the increased levels of pro-inflammatory cytokines, which are involved in the pathology of disease and the development of SARS-CoV-2-induced acute respiratory disease syndrome, the life-threatening form of this infection, are correlated with patient survival and disease severity. IL-33, a key cytokine involved in both innate and adaptive immune responses in mucosal organs, can increase airway inflammation, mucus secretion and Th2 cytokine synthesis in the lungs, following respiratory infections. Similar to cases of exposure to known respiratory virus infections, exposure to SARS-CoV-2 induces the expression of IL-33, correlating with T-cell activation and lung disease severity.
- IL-33
- COVID-19
- cytokine storm
- inflammation
1. Introduction
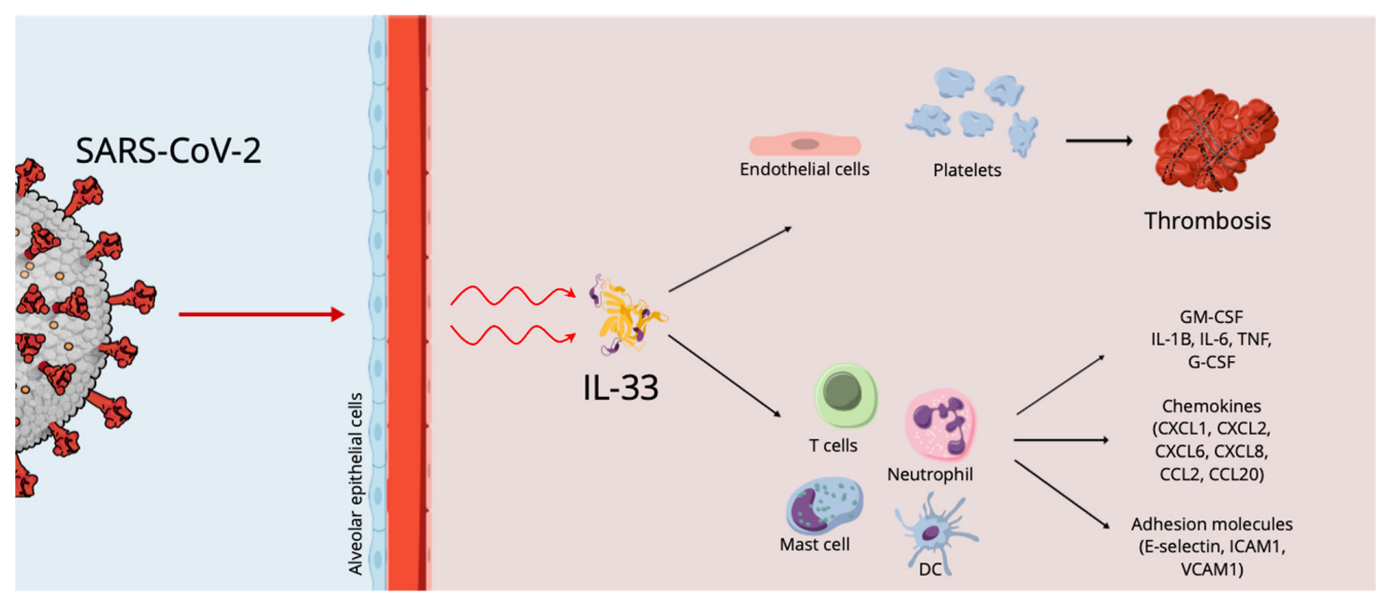
2. Cytokine Storm, IL-33 Effects and Thrombosis in COVID 19 Infection

This entry is adapted from the peer-reviewed paper 10.3390/ijms232314532
References
- Allegra, A.; Di Gioacchino, M.; Tonacci, A.; Musolino, C. Immunopathology of SARS-CoV-2 Infection: Immune Cells and Mediators, Prognostic Factors, and Immune-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 4782.
- Di Salvo, E.; Di Gioacchino, M.; Tonacci, A.; Casciaro, M.; Gangemi, S. Alarmins, COVID-19 and comorbidities. Ann. Med. 2021, 53, 777–785.
- Liang, Y.; Ge, Y.; Sun, J. IL-33 in COVID-19: Friend or foe? Cell. Mol. Immunol. 2021, 18, 1602–1604.
- Stanczak, M.A.; Sanin, D.E.; Apostolova, P.; Nerz, G.; Lampaki, D.; Hofmann, M.; Steimann, D.; Krohn-Grimberghe, M.; Thimme, R.; Mittler, G.; et al. IL-33 expression in response to SARS-CoV-2 correlates with seropositivity in COVID-19 convalescent individuals. Nat. Commun. 2021, 12, 2133.
- Markovic, S.S.; Jovanovic, M.; Gajovic, N.; Jurisevic, M.; Arsenijevic, N.; Jovanovic, M.; Jovanovic, M.; Mijailovic, Z.; Lukic, S.; Zornic, N.; et al. IL 33 Correlates With COVID-19 Severity, Radiographic and Clinical Finding. Front. Med. 2021, 8, 749569.
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069.
- Rubio-Sánchez, R.; Lepe-Balsalobre, E.; Viloria-Peñas, M. Prognostic factors for the severity of SARS-CoV-2 infection. Adv. Lab. Med. 2021, 2, 253–258.
- Meidaninikjeh, S.; Sabouni, N.; Marzouni, H.Z.; Bengar, S.; Khalili, A.; Jafari, R. Monocytes and macrophages in COVID-19: Friends and foes. Life Sci. 2021, 269, 119010.
- Miyazawa, M. Immunopathogenesis of SARS-CoV-2-induced pneumonia:lessons from influenza virus infection. Inflamm. Regen. 2020, 40, 39.
- Murdaca, G.; Paladin, F.; Tonacci, A.; Borro, M.; Greco, M.; Gerosa, A.; Isola, S.; Allegra, A.; Gangemi, S. Involvement of Il-33 in the Pathogenesis and Prognosis of Major Respiratory Viral Infections: Future Perspectives for Personalized Therapy. Biomedicines 2022, 10, 715.
- Zeng, Z.; Hong, X.Y.; Li, Y.; Chen, W.; Ye, G.; Li, Y.; Luo, Y. Serum-soluble ST2 as a novel biomarker reflecting inflammatory status and illness severity in patients with COVID-19. Biomark. Med. 2020, 14, 1619–1629.
- Cristinziano, L.; Poto, R.; Criscuolo, G.; Ferrara, A.L.; Galdiero, M.R.; Modestino, L.; Loffredo, S.; de Paulis, A.; Marone, G.; Spadaro, G.; et al. IL-33 and Superantigenic Activation of Human Lung Mast Cells Induce the Release of Angiogenic and Lymphangiogenic Factors. Cells 2021, 10, 145.
- Gaurav, R.; Anderson, D.R.; Radio, S.J.; Bailey, K.L.; England, B.R.; Mikuls, T.R.; Thiele, G.M.; Strah, H.M.; Romberger, D.J.; Wyatt, T.A.; et al. IL-33 Depletion in COVID-19 Lungs. Chest 2021, 160, 1656–1659.
- Opal, S.M. Interactions between coagulation and inflammation. Scand. J. Infect. Dis. 2003, 35, 545–554.
- Wifi, M.N.; Morad, M.A.; El Sheemy, R.; Abdeen, N.; Afify, S.; Abdalgaber, M.; Abdellatef, A.; Zaghloul, M.; Alboraie, M.; El-Kassas, M. Hemostatic system and COVID-19 crosstalk: A review of the available evidence. World J. Methodol. 2022, 12, 331–349.
- Nappi, F.; Nappi, P.; Gambardella, I.; Avtaar Singh, S.S. Thromboembolic Disease and Cardiac Thrombotic Complication in COVID-19: A Systematic Review. Metabolites 2022, 12, 889.
- Allegra, A.; Innao, V.; Allegra, A.G.; Musolino, C. Coagulopathy and thromboembolic events in patients with SARS-CoV-2 infection: Pathogenesis and management strategies. Ann. Hematol. 2020, 99, 1953–1965.
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649.
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329.
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587.
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13.
- Chaturvedi, S.; Braunstein, E.M.; Yuan, X.; Yu, J.; Alexander, A.; Chen, H.; Gavriilaki, E.; Alluri, R.; Streiff, M.B.; Petri, M.; et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood 2020, 135, 239–251.
- Gurbel, P.A.; Tantry, U.S. In Vitro Evidence for the Role of Cytokine Storm in the Generation of Stent Thrombosis in COVID-19 Patients. Cardiovasc. Revasc. Med. 2022, 35, 139–140.
- Demyanets, S.; Konya, V.; Kastl, S.P.; Kaun, C.; Rauscher, S.; Niessner, A.; Pentz, R.; Pfaffenberger, S.; Rychli, K.; Lemberger, C.E.; et al. Interleukin-33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2080–2089.
- Aoki, S.; Hayakawa, M.; Ozaki, H.; Takezako, N.; Obata, H.; Ibaraki, N.; Tsuru, T.; Tominaga, S.; Yanagisawa, K. ST2 gene expression is proliferation-dependent and its ligand, IL-33, induces inflammatory reaction in endothelial cells. Mol. Cell. Biochem. 2010, 335, 75–81.
- Choi, Y.S.; Choi, H.J.; Min, J.K.; Pyun, B.J.; Maeng, Y.S.; Park, H.; Kim, J.; Kim, Y.M.; Kwon, Y.G. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood 2009, 114, 3117–3126.
- Stojkovic, S.; Kaun, C.; Heinz, M.; Krychtiuk, K.A.; Rauscher, S.; Lemberger, C.E.; de Martin, R.; Gröger, M.; Petzelbauer, P.; Huk, I.; et al. Interleukin-33 induces urokinase in human endothelial cells--possible impact on angiogenesis. J. Thromb. Haemost. 2014, 12, 948–957.
- Demyanets, S.; Speidl, W.S.; Tentzeris, I.; Jarai, R.; Katsaros, K.M.; Farhan, S.; Krychtiuk, K.A.; Wonnerth, A.; Weiss, T.W.; Huber, K.; et al. Soluble ST2 and interleukin-33 levels in coronary artery disease: Relation to disease activity and adverse outcome. PLoS ONE. 2014, 9, e95055.
- Dhillon, O.S.; Narayan, H.K.; Khan, S.Q.; Kelly, D.; Quinn, P.A.; Squire, I.B.; Davies, J.E.; Ng, L.L. Pre-discharge risk stratification in unselected STEMI: Is there a role for ST2 or its natural ligand IL-33 when compared with contemporary risk markers? Int. J. Cardiol. 2013, 167, 2182–2188.
- Demyanets, S.; Tentzeris, I.; Jarai, R.; Katsaros, K.M.; Farhan, S.; Wonnerth, A.; Weiss, T.W.; Wojta, J.; Speidl, W.S.; Huber, K. An increase of interleukin-33 serum levels after coronary stent implantation is associated with coronary in-stent restenosis. Cytokine 2014, 67, 65–70.
- Stojkovic, S.; Kaun, C.; Basilio, J.; Rauscher, S.; Hell, L.; Krychtiuk, K.A.; Bonstingl, C.; de Martin, R.; Gröger, M.; Ay, C.; et al. Tissue factor is induced by interleukin-33 in human endothelial cells: A new link between coagulation and inflammation. Sci. Rep. 2016, 6, 25171.
- Stojkovic, S.; Thulin, Å.; Hell, L.; Thaler, B.; Rauscher, S.; Baumgartner, J.; Gröger, M.; Ay, C.; Demyanets, S.; Neumayer, C.; et al. IL-33 stimulates the release of procoagulant microvesicles from human monocytes and differentially increases tissue factor in human monocyte subsets. Thromb. Haemost. 2017, 117, 1379–1390.
- Barton, L.M.; Duval, E.; Stroberg, E.; Gosh, S.; Mukhopadhyay, S. COVID-19 autopsies, Oklahoma, U.S.A. Am. J. Clin. Pathol. 2020, 153, 725–733.
- Zeyda, M.; Wernly, B.; Demyanets, S.; Kaun, C.; Hammerle, M.; Hantusch, B.; Schranz, M.; Neuhofer, A.; Itariu, B.K.; Keck, M.; et al. Severe obesity increases adipose tissue expression of interleukin-33 and its receptor ST2, both predominantly detectable in endothelial cells of human adipose tissue. Int. J. Obes. 2013, 37, 658–665.
- Van Hinsbergh, V.W. Endothelium-role in regulation of coagulation and inflammation. Semin. Immunopathol. 2012, 34, 93–106.
- Demyanets, S.; Stojkovic, S.; Huber, K.; Wojta, J. The Paradigm Change of IL-33 in Vascular Biology. Int. J. Mol. Sci. 2021, 22, 13288.
- Chen, Z.; Luo, J.; Li, J.; Kim, G.; Stewart, A.; Huang, Y.; Wu, C. Intestinal IL-33 promotes platelet activity for neutrophil recruitment during acute inflammation. Blood 2022, 139, 1878–1891.
- Pariser, D.N.; Hilt, Z.T.; Ture, S.K.; Blick-Nitko, S.K.; Looney, M.R.; Cleary, S.J.; Roman-Pagan, E.; Saunders, J., II; Georas, S.N.; Veazey, J.; et al. Lung megakaryocytes are immune modulatory cells. J. Clin. Investig. 2021, 131, e137377.