 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jing Zhang | -- | 2568 | 2022-12-07 17:27:22 | | | |
| 2 | Lindsay Dong | Meta information modification | 2568 | 2022-12-08 02:54:39 | | |
Video Upload Options
Proliferating cells rely on DNA replication to ensure accurate genome duplication. Cancer cells, including breast cancer cells, exhibit elevated replication stress (RS) due to the uncontrolled oncogenic activation, loss of key tumor suppressors, and defects in the DNA repair machinery. This intrinsic vulnerability provides a great opportunity for therapeutic exploitation. An increasing number of drug candidates targeting RS in breast cancer are demonstrating promising efficacy in preclinical and early clinical trials.
1. Introduction
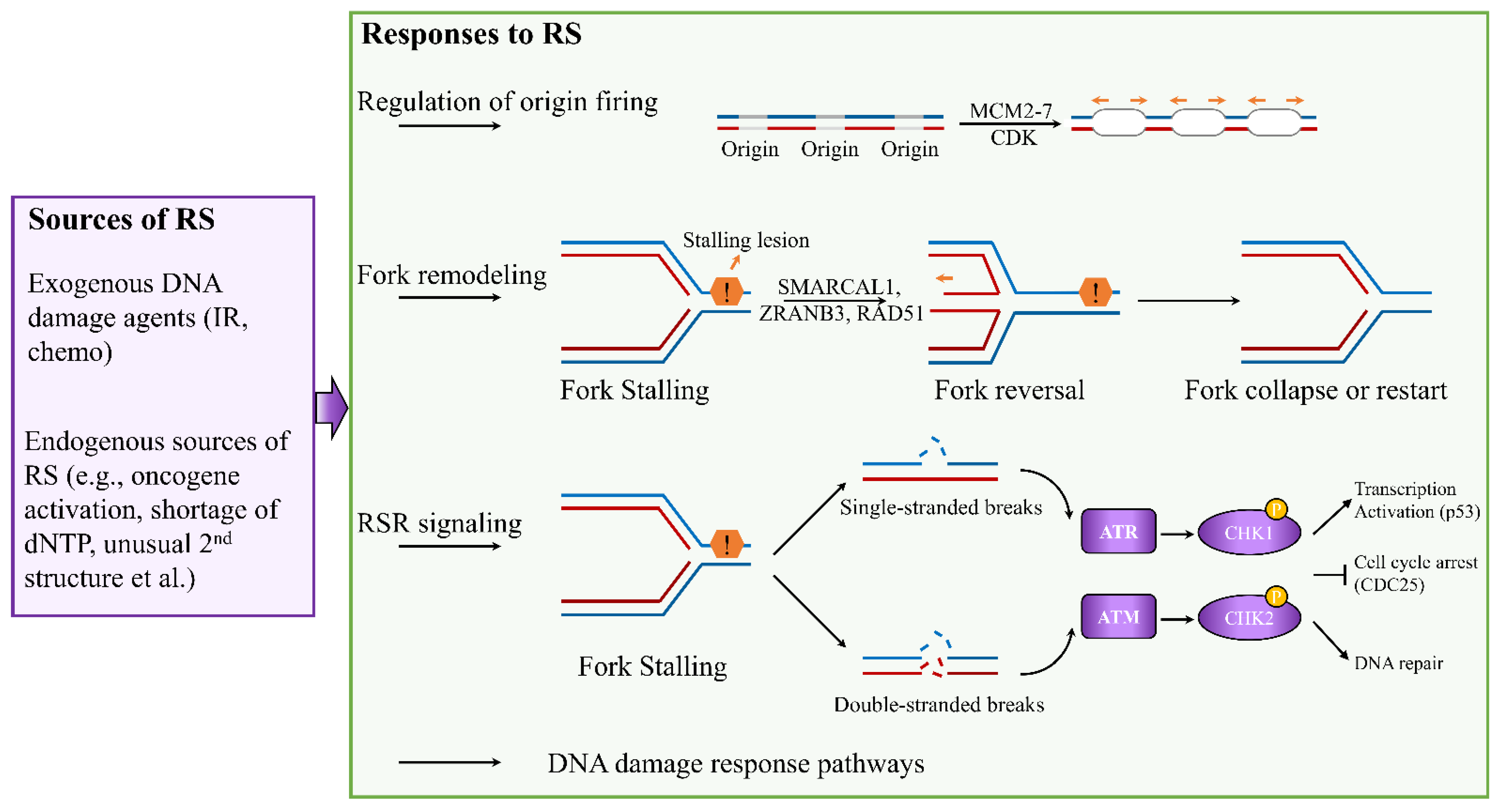
2. Mechanisms of the Cellular Response to Replication Stress and Rationale in Breast Cancer Therapy
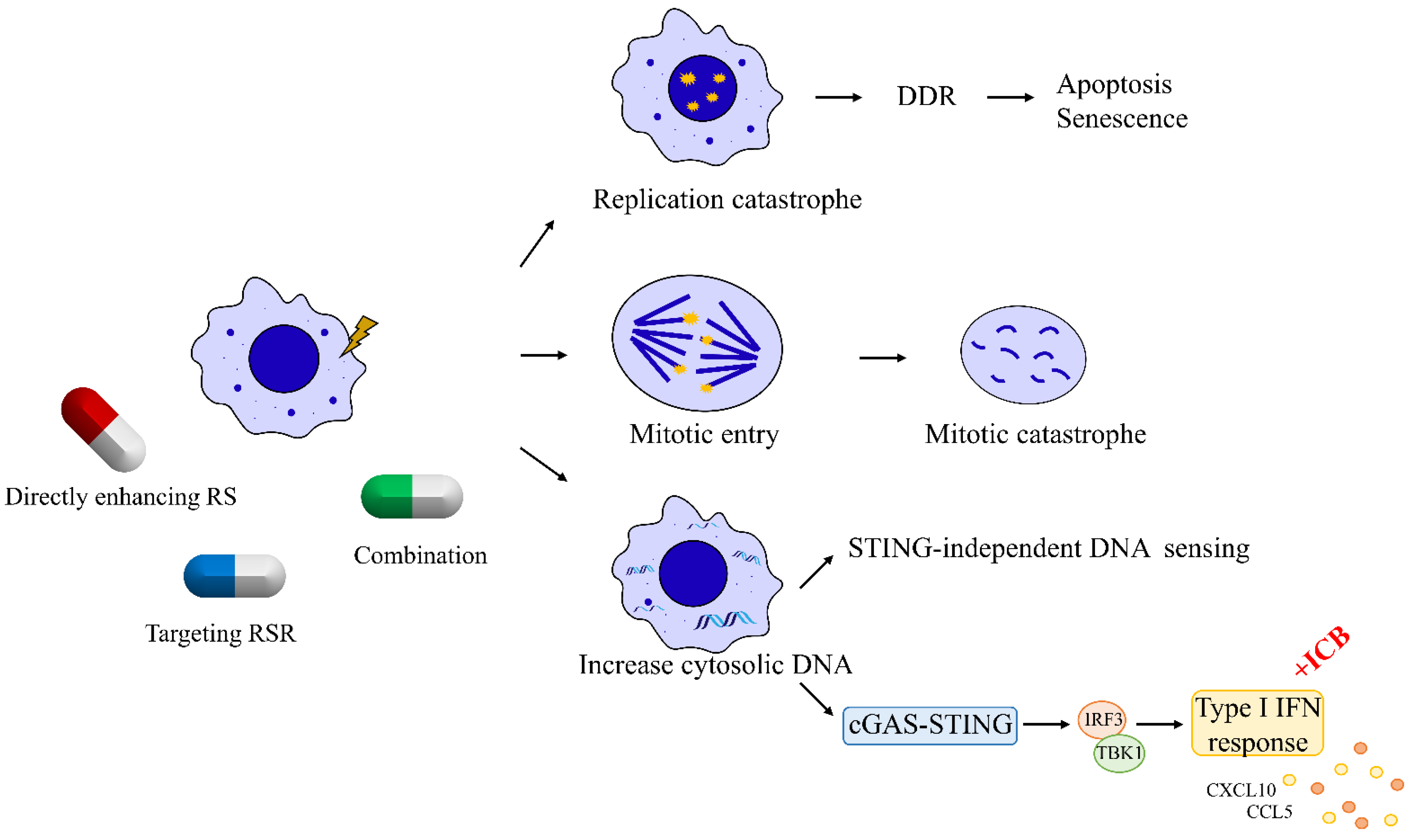
3. Therapeutic Strategies That Induce Replication Catastrophe in Breast Cancer
Since the persistence of replication stress is observed almost exclusively in cancer cells, enhancing replication stress can paradoxically lead to cell death by introducing further DNA damage in a catastrophic manner. Many traditional chemotherapeutic agents, acting by increasing the endogenous replication stress within breast cancer cells, have been developed and have shown antitumor activity. For instance, gemcitabine, a deoxycytidine analogue, leads to a delay in replication fork progression by inhibiting ribonucleotide reductase and by competing with dCTP for incorporation into newly synthesized DNA [43]. Another nucleoside analogue 5-fluorouracil, frequently used to treat breast cancer, functions mainly by inhibition of thymidylate synthetase to reduce the amount of thymidine for DNA replication [44][45]. TAS1553, a small-molecule subunit interaction inhibitor of ribonucleotide reductase, has shown antiproliferative activity in breast cancer cells by dramatically reducing the intracellular dATP pool and causing DNA replication stress [46].
4. Targeting Replication Stress Response Signaling
5. Emerging Combination Strategies of Immunotherapy with Agents Targeting RS
6. Predictive Biomarkers for Therapies Targeting Replication Stress
Optimal design of therapeutic strategies targeting RS requires reliable predictive biomarkers that can help to select, before the initiation of treatments, patients with breast cancer who would be most likely to benefit. Currently, there are a few biomarkers that have been identified to predict the response of patients with breast cancer to drugs enhancing RS. For example, Birkbak et al. reported that the expression levels of the BLM and FANCI genes are potential biomarkers that predict response of TNBC to platinum-based therapy [75].
On the basis of positive outcomes in clinical trials, PARP inhibitors olaparib and talazoparib are approved as monotherapies for the treatment of patients with germline BRCA-mutated, HER2-negative advanced or metastatic breast cancer [76][77][78]. However, PARPi resistance has proved to be a major problem in the clinic [79]. The mechanisms underlying this resistance have been characterized, and restoration of replication fork stability is one of major mechanisms for PARPi resistance [79]. Collectively, these studies suggest that, owing to the complexity of tumor microenvironment, single biomarkers are insufficient to accurately predict clinical outcomes with therapies targeting RS, and a combination of genetic deficiencies will be required to deliver a sufficient degree of sensitivity to drugs targeting RS.
One key question remains to be addressed, when identifying biomarkers that indicate a dependency on the RSR and a likely response to inhibitors targeting key RSR kinases, is whether the clinical biopsy taken at initial diagnosis still indicates the level of RSR dependence in the tumor to be treated. Therefore, genetic analysis of clinical samples obtained at different timepoints, i.e., pre- and post-treatment tumor samples, may represent an important approach for understanding the dynamic changes of response to agents targeting RSR, and an additional goal would be the identification of more reliable predictive biomarkers that can help to select patients prior to therapy.
References
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374.
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9.
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109.
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196.
- McGrail, D.J.; Lin, C.C.; Dai, H.; Mo, W.; Li, Y.; Stephan, C.; Davies, P.; Lu, Z.; Mills, G.B.; Lee, J.S.; et al. Defective Replication Stress Response Is Inherently Linked to the Cancer Stem Cell Phenotype. Cell Rep. 2018, 23, 2095–2106.
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289.
- Ngoi, N.Y.L.; Pham, M.M.; Tan, D.S.P.; Yap, T.A. Targeting the replication stress response through synthetic lethal strategies in cancer medicine. Trends Cancer 2021, 7, 930–957.
- Dobbelstein, M.; Sørensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423.
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637.
- Dapic, V.; Carvalho, M.A.; Monteiro, A.N.A. Breast Cancer Susceptibility and the DNA Damage Response. Cancer Control 2005, 12, 127–136.
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355.
- Osborn, A.J.; Elledge, S.J.; Zou, L. Checking on the fork: The DNA-replication stress-response pathway. Trends Cell Biol. 2002, 12, 509–516.
- Nazareth, D.; Jones, M.J.; Gabrielli, B. Everything in Moderation: Lessons Learned by Exploiting Moderate Replication Stress in Cancer. Cancers 2019, 11, 1320.
- Rajamanickam, S.; Park, J.; Bates, K.; Timilsina, S.; Eedunuri, V.; Onyeagucha, B.; Subbarayalu, P.; Abdelfattah, N.; Jung, K.; Favours, E.; et al. Abstract P6-06-04: Targeting replication stress in triple negative breast cancer treatment regimen: An emerging approach. Cancer Res. 2018, 78, P6-06-04.
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751.
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120.
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818.
- Técher, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017, 18, 535–550.
- Liu, W.; Krishnamoorthy, A.; Zhao, R.; Cortez, D. Two replication fork remodeling pathways generate nuclease substrates for distinct fork protection factors. Sci. Adv. 2020, 6, eabc3598.
- Quinet, A.; Lemaçon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833.
- Saxena, S.; Zou, L. Hallmarks of DNA replication stress. Mol. Cell 2022, 82, 2298–2314.
- Abe, S.; Yamamoto, K.; Kurata, M.; Abe-Suzuki, S.; Horii, R.; Akiyama, F.; Kitagawa, M. Targeting MCM2 function as a novel strategy for the treatment of highly malignant breast tumors. Oncotarget 2015, 6, 34892–34909.
- Juríková, M.; Danihel, Ľ.; Polák, Š.; Varga, I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016, 118, 544–552.
- Issac, M.S.M.; Yousef, E.; Tahir, M.R.; Gaboury, L.A. MCM2, MCM4, and MCM6 in Breast Cancer: Clinical Utility in Diagnosis and Prognosis. Neoplasia 2019, 21, 1015–1035.
- Mahadevappa, R.; Neves, H.; Yuen, S.M.; Bai, Y.; McCrudden, C.M.; Yuen, H.F.; Wen, Q.; Zhang, S.D.; Kwok, H.F. The prognostic significance of Cdc6 and Cdt1 in breast cancer. Sci. Rep. 2017, 7, 985.
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Técher, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881.e7.
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.-W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8.
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387.
- Ding, X.; Ray Chaudhuri, A.; Callen, E.; Pang, Y.; Biswas, K.; Klarmann, K.D.; Martin, B.K.; Burkett, S.; Cleveland, L.; Stauffer, S.; et al. Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun. 2016, 7, 12425.
- Wu, X. Replication Stress Response Links RAD52 to Protecting Common Fragile Sites. Cancers 2019, 11, 1467.
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859.
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548.
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45.
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P.; et al. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair 2014, 21, 131–139.
- Shechter, D.; Costanzo, V.; Gautier, J. ATR and ATM regulate the timing of DNA replication origin firing. Nat. Cell Biol. 2004, 6, 648–655.
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167.
- Mailand, N.; Falck, J.; Lukas, C.; Syljuâsen, R.G.; Welcker, M.; Bartek, J.; Lukas, J. Rapid destruction of human Cdc25A in response to DNA damage. Science 2000, 288, 1425–1429.
- Ubhi, T.; Brown, G.W. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res. 2019, 79, 1730–1739.
- Schoonen, P.M.; Guerrero Llobet, S.; van Vugt, M. Replication stress: Driver and therapeutic target in genomically instable cancers. Adv. Protein Chem. Struct. Biol. 2019, 115, 157–201.
- Zhang, J.; Shih, D.J.H.; Lin, S.-Y. Role of DNA repair defects in predicting immunotherapy response. Biomark. Res. 2020, 8, 23.
- Coquel, F.; Silva, M.-J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.-L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61.
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017, 31, 353–369.
- Merriman, R.L.; Hertel, L.W.; Schultz, R.M.; Houghton, P.J.; Houghton, J.A.; Rutherford, P.G.; Tanzer, L.R.; Boder, G.B.; Grindey, G.B. Comparison of the antitumor activity of gemcitabine and ara-C in a panel of human breast, colon, lung and pancreatic xenograft models. Investig. New Drugs 1996, 14, 243–247.
- Cameron, D.A.; Gabra, H.; Leonard, R.C. Continuous 5-fluorouracil in the treatment of breast cancer. Br. J. Cancer 1994, 70, 120–124.
- Ponce-Cusi, R.; Calaf, G.M. Apoptotic activity of 5-fluorouracil in breast cancer cells transformed by low doses of ionizing α-particle radiation. Int. J. Oncol. 2016, 48, 774–782.
- Ueno, H.; Hoshino, T.; Yano, W.; Tsukioka, S.; Suzuki, T.; Hara, S.; Ogino, Y.; Chong, K.T.; Suzuki, T.; Tsuji, S.; et al. TAS1553, a small molecule subunit interaction inhibitor of ribonucleotide reductase, exhibits antitumor activity by causing DNA replication stress. Commun. Biol. 2022, 5, 571.
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320.
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120.
- Promonet, A.; Padioleau, I.; Liu, Y.; Sanz, L.; Biernacka, A.; Schmitz, A.-L.; Skrzypczak, M.; Sarrazin, A.; Mettling, C.; Rowicka, M.; et al. Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat. Commun. 2020, 11, 3940.
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst.) 2018, 71, 172–176.
- Michelena, J.; Lezaja, A.; Teloni, F.; Schmid, T.; Imhof, R.; Altmeyer, M. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat. Commun. 2018, 9, 2678.
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331.e3.
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 2021, 81, 3128–3144.e7.
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284.
- Han, T.; Goralski, M.; Capota, E.; Padrick, S.B.; Kim, J.; Xie, Y.; Nijhawan, D. The antitumor toxin CD437 is a direct inhibitor of DNA polymerase α. Nat. Chem. Biol. 2016, 12, 511–515.
- Shao, Z.M.; Dawson, M.I.; Li, X.S.; Rishi, A.K.; Sheikh, M.S.; Han, Q.X.; Ordonez, J.V.; Shroot, B.; Fontana, J.A. p53 independent G0/G1 arrest and apoptosis induced by a novel retinoid in human breast cancer cells. Oncogene 1995, 11, 493–504.
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103.
- Chen, Y.H.; Jones, M.J.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J., III; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338.
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595.
- Roulston, A.; Zimmermann, M.; Papp, R.; Skeldon, A.; Pellerin, C.; Dumas-Bérube, É.; Dumais, V.; Dorich, S.; Fader, L.D.; Fournier, S.; et al. RP-3500: A Novel, Potent, and Selective ATR Inhibitor that is Effective in Preclinical Models as a Monotherapy and in Combination with PARP Inhibitors. Mol. Cancer Ther. 2022, 21, 245–256.
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, e1013–e1824.
- Pitts, T.M.; Simmons, D.M.; Bagby, S.M.; Hartman, S.J.; Yacob, B.W.; Gittleman, B.; Tentler, J.J.; Cittelly, D.; Ormond, D.R.; Messersmith, W.A.; et al. Wee1 Inhibition Enhances the Anti-Tumor Effects of Capecitabine in Preclinical Models of Triple-Negative Breast Cancer. Cancers 2020, 12, 719.
- Sand, A.; Piacsek, M.; Donohoe, D.L.; Duffin, A.T.; Riddell, G.T.; Sun, C.; Tang, M.; Rovin, R.A.; Tjoe, J.A.; Yin, J. WEE1 inhibitor, AZD1775, overcomes trastuzumab resistance by targeting cancer stem-like properties in HER2-positive breast cancer. Cancer Lett. 2020, 472, 119–131.
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 510–519.
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage–Inducing or Repair–Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38.
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065.
- Nanda, R.; Chow, L.Q.M.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467.
- Emens, L.A.; Braiteh, F.S.; Cassier, P.; Delord, J.-P.; Eder, J.P.; Fasso, M.; Xiao, Y.; Wang, Y.; Molinero, L.; Chen, D.S.; et al. Abstract 2859: Inhibition of PD-L1 by MPDL3280A leads to clinical activity in patients with metastatic triple-negative breast cancer (TNBC). Cancer Res. 2015, 75, 2859.
- Lin, Y.-L.; Pasero, P. Replication stress: From chromatin to immunity and beyond. Curr. Opin. Genet. Dev. 2021, 71, 136–142.
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199.
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.-P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920.
- McGrail, D.J.; Pilié, P.G.; Dai, H.; Lam, T.N.A.; Liang, Y.; Voorwerk, L.; Kok, M.; Zhang, X.H.-F.; Rosen, J.M.; Heimberger, A.B.; et al. Replication stress response defects are associated with response to immune checkpoint blockade in nonhypermutated cancers. Sci. Transl. Med. 2021, 13, eabe6201.
- Sun, L.L.; Yang, R.Y.; Li, C.W.; Chen, M.K.; Shao, B.; Hsu, J.M.; Chan, L.C.; Yang, Y.; Hsu, J.L.; Lai, Y.J.; et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am. J. Cancer Res. 2018, 8, 1307–1316.
- Asleh, K.; Riaz, N.; Cheng, A.S.; Gao, D.; Leung, S.C.Y.; Anurag, M.; Nielsen, T.O. Proteomics-derived basal biomarker DNA-PKcs is associated with intrinsic subtype and long-term clinical outcomes in breast cancer. NPJ Breast Cancer 2021, 7, 114.
- Proctor, M.; Gonzalez Cruz, J.L.; Daignault-Mill, S.M.; Veitch, M.; Zeng, B.; Ehmann, A.; Sabdia, M.; Snell, C.; Keane, C.; Dolcetti, R.; et al. Targeting Replication Stress Using CHK1 Inhibitor Promotes Innate and NKT Cell Immune Responses and Tumour Regression. Cancers 2021, 13, 3733.
- Birkbak, N.J.; Li, Y.; Pathania, S.; Greene-Colozzi, A.; Dreze, M.; Bowman-Colin, C.; Sztupinszki, Z.; Krzystanek, M.; Diossy, M.; Tung, N.; et al. Overexpression of BLM promotes DNA damage and increased sensitivity to platinum salts in triple-negative breast and serous ovarian cancers. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 903–909.
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533.
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763.
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282.
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834.

