Proliferating cells rely on DNA replication to ensure accurate genome duplication. Cancer cells, including breast cancer cells, exhibit elevated replication stress (RS) due to the uncontrolled oncogenic activation, loss of key tumor suppressors, and defects in the DNA repair machinery. This intrinsic vulnerability provides a great opportunity for therapeutic exploitation. An increasing number of drug candidates targeting RS in breast cancer are demonstrating promising efficacy in preclinical and early clinical trials.
1. Introduction
DNA replication in eukaryotic cells is a multifaceted process, which depends on the activation of numerous signaling pathways to accurately replicate the genome [
1]. This process is constantly challenged by events of endogenous or exogenous origin that impede the rate and fidelity of DNA synthesis, thus affecting the integrity of genome. These events, collectively termed replication stress (RS), include DNA lesions such as DNA single-stranded or double-stranded breaks, unusual DNA secondary structures, RNA–DNA hybrids, deficiencies in nucleotide levels, oncogene activation, chromatin inaccessibility and limitation of essential replication factors [
2]. In response to RS, cells elicit the DNA damage response (DDR) and subsequently inhibition of cell-cycle progression, which is known as the replication stress response (RSR). The purpose of RSR is to slow DNA synthesis and replication to allow time for DNA repair [
3]. The RSR is primarily coordinated by two signaling cascades: ataxia telangiectasia and Rad3-related (ATR)–checkpoint kinase 1 (CHK1) pathway and ataxia telangiectasia mutated (ATM)–checkpoint kinase 2 (CHK2) pathway [
4,
5,
6,
7]. Cells rely on these coordinated pathways to prevent mitosis in the presence of DNA damage [
8]. Defects in RSR allow for high levels of DNA damage and genomic instability that not only alter gene function, but also lead to continuous proliferation, ultimately provoking carcinogenesis and tumor progression [
9,
10,
11,
12,
13].
Breast cancer is one of the most frequently diagnosed neoplasms worldwide, with one-third of these patients subsequently dying of this disease. In this heterogeneous disease, an important cause of replication stress is overexpression or constitutive activation of oncogenes, loss of key tumor suppressors, and defects in the DNA repair machinery [
2]. Over the past decades, treatments for breast cancer have advanced considerably with effective therapies targeting the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2 (HER2). However, patients with triple-negative breast cancer (TNBC), i.e., estrogen receptor-negative, progesterone receptor-negative and HER2-negative, fail to benefit from these treatments. Notably, it was reported that TNBC has high levels of RS due to the activation of various oncogenes and germline BRCA mutations or “BRCAness” in the absence of BRCA mutations [
14]. BRCAness is defined as a defect in double-strand break repair (DSBR) via homologous recombination repair (HRR) [
15,
16]. In the HRR process, recombinase Rad51 interacts with BRCA1 and BRCA2 to perform the search for homologous DNA sequences [
16,
17].
2. Mechanisms of the Cellular Response to Replication Stress and Rationale in Breast Cancer Therapy
Accurate DNA replication is critical to ensure genomic integrity. The fidelity of this process is often challenged by RS, leading to altered replication fork progression and generation of DNA breaks [
2,
22]. Depending on the types of RS, cells activate four major responses to ensure accurate genome duplication: (1) regulation of origin firing, (2) remodeling of replication forks, (3) activation of replication stress response signaling, and (4) deployment of DNA damage response pathways (
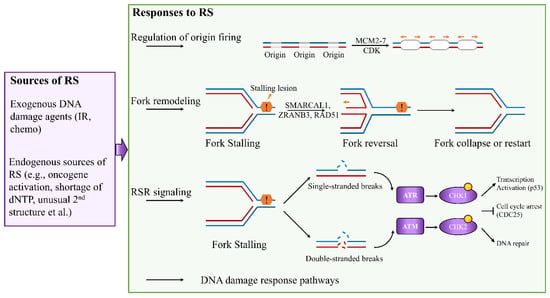
Figure 1) [
23,
24,
25].
Figure 1. Cellular responses to DNA replication stress. Depending on the types of RS, cells activate numerous responses to ensure accurate completion of genome duplication, including regulation of origin firing, replication fork remodeling, activation of RSR signaling, and involvement of DNA damage response pathways.
Eukaryotic genomes are duplicated exactly once during the S phases of each cell cycle. To control replication initiation, the replicative DNA helicase minichromosome maintenance complex 2–7 (MCM2–7) is loaded at replication origins during G1 phase, and activated only during the S phase [
6]. This process is called licensing. Activation of licensed origins, which is known as origin firing, requires the activities of cyclin-dependent kinase (CDK) [
6]. Deregulation of origin activation can generate replication stress in breast cancer. For example, high levels of MCM2 are associated with poor survival in patients with breast cancer [
26,
27]. Furthermore, Issac et al. have showed that increased protein expression of MCM2, MCM4, and MCM6 is associated with luminal B, HER2-positive, and TNBC [
28]. These MCM proteins may serve as potential treatment targets for breast cancer patients. Unscheduled replication is another source of replication stress in breast cancer. This occurs when the timing of origin activation is altered, leading to DNA regions replicating more than once in one cell cycle or an increase in origin firing in early S phase [
6]. Unscheduled replication occurs under the upregulation of DNA replication factor CDT1 and cell division control protein 6 (CDC6). High levels of CDT1 and CDC6 are associated with poorer survival in the breast cancer patients, suggesting that CDT1 and CDC6 are potential therapeutic targets for treatment of breast cancer [
29]. To date, the mechanism that regulate origin firing remain largely unknown, and few drugs targeting these key proteins are in the clinical trials. Thus, better understanding the regulation of the origin firing can provide novel therapeutic strategies in the treatment of breast cancer.
Replication fork remodeling is a common response to RS, which involves unwinding of newly synthesized strands and annealing of parental strands. In this remodeling process, stalled forks are converted into four-way junctions to facilitate DNA damage repair [
24]. Many key factors involved in reversed fork restart have been identified, including ZRANB3 and SMARCAL1 in reversed fork formation, and BRCA1 and BRCA2 in reversed fork protection [
30,
31]. RAD51, well-known for catalyzing strand invasion in homologous recombination (HR) repair of DNA double-strand breaks, also plays an important role in regulating replication fork reversal [
23]. Furthermore, PARP1 has been linked to recruitment of MRE11 to stalled forks and regulation of fork restart to restore replication fork stability [
32,
33]. Similar to PARP1, RAD52 can promote the recruitment of MRE11 to stalled replication forks and fork degradation [
34,
35].
To cope with RS, cells can also rely on RSR cascade to arrest the cell cycle, protect stalled forks, and allow time to repair the replication fork (
Figure 1). RS can perturb the coupling between the replicative helicase and polymerases, resulting in the uncoupling of leading- and lagging-strand synthesis, which generates double-strand breaks (DSBs) and/or single-strand DNA (ssDNA) gaps [
6]. DSBs primarily trigger activation of ATM and DNA-dependent protein kinase (DNA-PK), whereas ssDNA coated with replication protein A (RPA) activates ATR via ATR-interacting protein (ATRIP) [
39,
40,
41]. ATR and ATM are apical checkpoint kinases, which regulate the cellular response to replication fork blockage and DNA damage [
42]. These kinases activate the effector checkpoint kinases CHK1 and CHK2, respectively, and they regulate the timing of replication origin firing independently of DNA damage [
42]. Activation of the ATR–CHK1 pathway leads to cell-cycle arrest by inactivating the cell division cycle 25 (CDC25) phosphatase family or through WEE1 kinase [
43,
44].
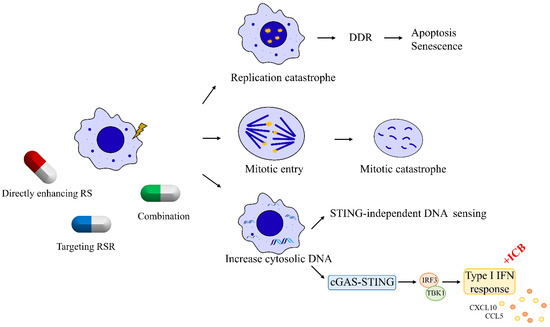
Failure to remove replication stressors due to the loss of replication stress response and repair proteins is a prominent feature of tumor cells. Because of this key feature that distinguishes cancer cells from normal cells, cancer cells harboring replication stress can be targeted through three major mechanisms (
Figure 2). First, cancer cells can be pushed toward cell death by enhancing replication stress to induce replication catastrophe [
21,
49]. Second, cancer cells can be targeted by inhibiting the key kinases of the RSR cascade that cells depend upon to survive, such as ATR, ATM, CHK1, WEE1, and DNA-PK. Inhibition of these key mediators ultimately promotes the premature entry of tumor cells into mitosis, inducing mitotic catastrophe [
7,
49]. Third, replication stress due to chemotherapies or impaired DNA repair factors induces DNA release from the nucleus to cytoplasm. These DNA fragments trigger cytosolic DNA sensing and interferon signaling, which subsequently prime tumor for immune therapies [
7,
50,
51,
52].
Figure 2. Targeting DNA replication stress in breast cancer. Breast cancer cells harboring RS can be targeted through three major mechanisms. First, RS can be harnessed in breast cancer cells with high intrinsic RS to induce replication catastrophe. Second, breast cancer cells can be targeted by abrogating their G2–M cell-cycle checkpoint to induce mitotic catastrophe. Third, breast cancer cells with high intrinsic RS exhibit high levels of cytosolic DNA leaking from nucleus, which can trigger interferon signaling to prime tumor for immune therapies.
3. Therapeutic Strategies That Induce Replication Catastrophe in Breast Cancer
Since the persistence of replication stress is observed almost exclusively in cancer cells, enhancing replication stress can paradoxically lead to cell death by introducing further DNA damage in a catastrophic manner. Many traditional chemotherapeutic agents, acting by increasing the endogenous replication stress within breast cancer cells, have been developed and have shown antitumor activity. For instance, gemcitabine, a deoxycytidine analogue, leads to a delay in replication fork progression by inhibiting ribonucleotide reductase and by competing with dCTP for incorporation into newly synthesized DNA [54]. Another nucleoside analogue 5-fluorouracil, frequently used to treat breast cancer, functions mainly by inhibition of thymidylate synthetase to reduce the amount of thymidine for DNA replication [55,56]. TAS1553, a small-molecule subunit interaction inhibitor of ribonucleotide reductase, has shown antiproliferative activity in breast cancer cells by dramatically reducing the intracellular dATP pool and causing DNA replication stress [57].
Unlike nucleoside analogues, which reduce dNTP pools to direct inhibition of DNA synthesis, alkylating agents and platinum-containing compounds increase the replication stress in breast cancer cells by directly modifying DNA through attacking the DNA bases forming covalent DNA adducts [
83,
84]. The formation of DNA adducts interferes with the progression of DNA polymerases, which results in delay replication fork progression. In addition to these RS inducers, topoisomerase inhibitors can increase replication stress by promoting R-loop formation, leading to global replication fork slowdown [
85]. Another class of replication stress inducing agents that target breast cancer cells by directly inducing DNA damage is represented by poly(ADP-ribose) polymerase (PARP) inhibitors. PARP inhibitors are believed to interfere with the replication machinery and promote fork collapse by trapping PARP on DNA [
86,
87]. Additionally, PARP inhibitors cause an accumulation of single-strand breaks (SSBs) or Okazaki fragment processing [
88,
89]. More recently, PARP inhibitors were reported to increase the speed of fork elongation and amplify the replication stress in breast cancer cells [
90].
Given that DNA polymerases play key roles in replication stress sensing upstream of ATR, inhibitors targeting DNA polymerases are a novel and attractive class of drugs that are in preclinical development. CD437, targeting POLA1 which encodes DNA polymerase α, has been shown to induce apoptosis in breast cancer cells but not in normal cells [
67,
91].
4. Targeting Replication Stress Response Signaling
If breast cancer cells with high levels of replicative lesions do not initiate cell death, they likely rely on RSR to provide sufficient time to deal with such lesions. The main RSR mediators for inducing cell-cycle delay or protecting stalled forks could, therefore, be promising targets (
Table 1). Given the central role of ATR in preventing replication fork collapse, ATR inhibition initiates widespread DNA synthesis from dormant replication origins, generating ssDNA to exhaust the cellular pools of RPA [
98,
99]. ATR inhibitors (ATRi), such as AZD6738, BAY1895344, and M6620, have shown antitumor activity in preclinical and clinical studies [
100]. Additional agent RP-3500 targeting ATR in breast cancer is currently under clinical phase 1 trial [
72]. CHK1, the key downstream effector protein of the ATR response, plays an important role in triggering the S-phase checkpoint upon replication stress and preventing premature entry into mitosis. Currently, two CHK1 inhibitors, prexasertib [
10In addition to these two key players, the RSR also involves other important kinases, including WEE1, DNA-PK, and ATM, to arrest the cell cycle, protect stalled forks, and allow time for replication fork repair. For instance, the activation of ATR–CHK1 signaling can delay cell-cycle progression through WEE1 kinase [
43]. A small-molecular inhibitor of WEE1 kinase, AZD1775, can increase unscheduled origin firings, leading to replication fork stalling and driving HER2-positive or TNBC breast cells into unscheduled mitosis [
102,
103].
Limitations associated with the use of these inhibitors, such as excessive toxicity at effective doses and resistance to the treatment through compensatory pathways, promote the development of combination therapies. ATR and CHK1 inhibitors synergize with compounds that induce replication stress in breast cancers, including nucleoside analogues, platinum-based agents, and PARP inhibitors [
107,
108]. Additionally, a preclinical study showed that DNA-PK inhibitor AZD7648 enhances the efficiency of doxorubicin and PARP inhibitors in breast cancer cell lines and TNBC patient-derived xenograft models [
109]. Clinical trials evaluating the antitumor efficacy of combining DNA-PK inhibitor and CHK1 inhibitor in breast cancer are ongoing (NCT04032080 and NCT02124148).
5. Emerging Combination Strategies of Immunotherapy with Agents Targeting RS
Immunotherapy, especially immune checkpoint blockade (ICB), is emerging as a new treatment modality in breast cancer. Monotherapy using antibodies against programmed death-1 (PD-1) and programmed death ligand-1 (PD-L1) reported objective response rates (ORRs) of around 10% to 20% in patients with metastatic breast cancer [
112,
113]. The lack of response of patients with breast cancer to immunotherapy has directed research toward novel combination therapeutic strategies aimed at transforming a higher proportion of non-responders into responders
Preclinical studies have shown the link between immune response and pathways involved in RS. Accumulating evidence indicates that RS triggers the immune response through the accumulation of cytosolic DNA derived from the nucleus and the activation of DNA sensing pathways [
21,
114]. For instance, S-phase-specific DNA damage in breast cancer was identified to be associated with increased T-cell infiltration and PD-L1 expression in a STING-dependent manner [
115]. Additionally, Diamond et al. showed that irradiated breast cancer cells transferred cytosolic dsDNA to dendritic cells and stimulated dendritic cell upregulation of costimulatory molecules and STING-dependent activation of IFN signaling [
116]. Recently, a preclinical study reported that induction of RSR defects by CHK1/2 inhibition improved ICB response in murine breast cancer models [
117]. Furthermore, other recent studies have illustrated how key RSR members including ATR, CHK1, and DNA-PK regulate immune response in breast cancer [
118,
119,
120]. These studies suggested that targeting RS would amplify the response of patients with breast cancer to ICB.
6. Predictive Biomarkers for Therapies Targeting Replication Stress
Optimal design of therapeutic strategies targeting RS requires reliable predictive biomarkers that can help to select, before the initiation of treatments, patients with breast cancer who would be most likely to benefit. Currently, there are a few biomarkers that have been identified to predict the response of patients with breast cancer to drugs enhancing RS. For example, Birkbak et al. reported that the expression levels of the BLM and FANCI genes are potential biomarkers that predict response of TNBC to platinum-based therapy [128].
On the basis of positive outcomes in clinical trials, PARP inhibitors olaparib and talazoparib are approved as monotherapies for the treatment of patients with germline BRCA-mutated, HER2-negative advanced or metastatic breast cancer [130,131,132]. However, PARPi resistance has proved to be a major problem in the clinic [133]. The mechanisms underlying this resistance have been characterized, and restoration of replication fork stability is one of major mechanisms for PARPi resistance [133]. Collectively, these studies suggest that, owing to the complexity of tumor microenvironment, single biomarkers are insufficient to accurately predict clinical outcomes with therapies targeting RS, and a combination of genetic deficiencies will be required to deliver a sufficient degree of sensitivity to drugs targeting RS.
One key question remains to be addressed, when identifying biomarkers that indicate a dependency on the RSR and a likely response to inhibitors targeting key RSR kinases, is whether the clinical biopsy taken at initial diagnosis still indicates the level of RSR dependence in the tumor to be treated. Therefore, genetic analysis of clinical samples obtained at different timepoints, i.e., pre- and post-treatment tumor samples, may represent an important approach for understanding the dynamic changes of response to agents targeting RSR, and an additional goal would be the identification of more reliable predictive biomarkers that can help to select patients prior to therapy.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10112775