 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Petar Todorov Todorov | -- | 5789 | 2022-12-01 17:34:07 | | | |
| 2 | Petar Todorov Todorov | -3 word(s) | 5786 | 2022-12-04 10:50:24 | | | | |
| 3 | Rita Xu | -46 word(s) | 5740 | 2022-12-05 03:03:00 | | |
Video Upload Options
The endogenous hemorphins are bioactive peptides with activity on opioid receptors. Several research teams have synthesized, characterized, and pharmacologically evaluated synthetic hemorphin analogs containing unusual amino acids, D-amino acids, α-aminophosphonic acids, and their derivatives. Research focuses on the structure-activity relationship analysis, details on specific methods for their characterization, and the advantage of synthetic hemorphin analogs compared to endogenous peptides as potent biologically active compounds with a complex mechanism of action.
1. Introduction
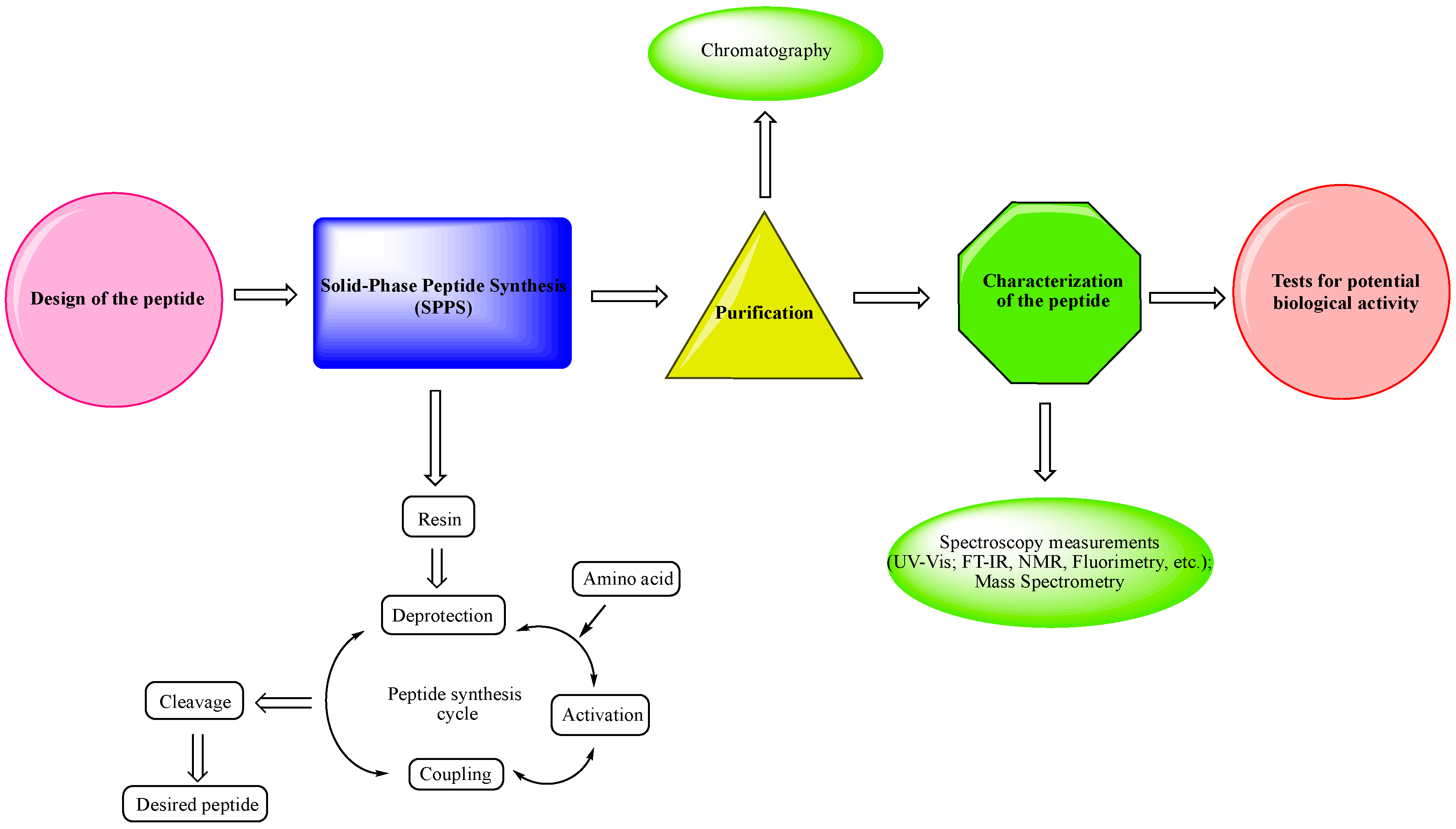
2. Chemistry and Biology of Synthetic Hemorphin Analogs Containing Non-Natural Amino Acids
- -
-
Design of the peptide—planning of the desired peptide compound with expected biological activity, what modifications to be made, in which part of the molecule to be made, what properties researchers expect to obtain, etc.
- -
-
Choice of a reliable method used to obtain the desired peptide—peptide synthesis in solution or solid-phase peptide synthesis (SPPS). The solid-phase peptide synthesis by the Fmoc-strategy is the most widespread and acceptable method due to the number of its advantages, including reduced reaction time for creating a peptide bond; quantitative progression of condensation reactions; the easy removal of excess reagents and solvents by washing the peptidyl-resin; minimal losses when receiving the final product.
- -
-
The synthesized peptide must be purified using chromatography (the most used is reversed-phase high-performance liquid chromatography (RP-HPLC)).
- -
-
Followed by the complete characterization of the peptide using modern instrumental methods and techniques: spectroscopy measurements (UV-Vis; FT-IR, NMR, fluorimetry, etc.); mass spectrometry.
- -
-
Screening tests for potential biological activity.
| № | Abbreviations Given in Articles | Peptide | Molecular Formula | Biological Activity, Reference |
|---|---|---|---|---|
| Hemorphin-4 analogs | ||||
| 1 | P4-1 | Tyr-Ac5c-Trp-Thr-NH2 | C30H38N6O6 | anticonvulsant activity, [44] |
| 2 | P4-2 | Tyr-Ac6c-Trp-Thr-NH2 | C31H40N6O6 | anticonvulsant activity, [44] |
| 3 | P4-3 | Aaa-Tyr-Pro-Trp-Thr-NH2 | C40H50N6O7 | anticonvulsant activity, [44] |
| 4 | P4-4 | Aaa-Tyr-Ac5c-Trp-Thr-NH2 | C41H52N6O7 | anticonvulsant activity, [44] |
| 5 | P4-5 | Aaa-Tyr-Ac6c-Trp-Thr-NH2 | C42H54N6O7 | anticonvulsant activity, [44] |
| 6 | Dm-4 |  |
C36H44N8O9 | anticonvulsant activity, [45] |
| 7 | Ph-4 | 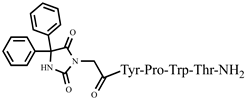 |
C46H48N8O9 | anticonvulsant activity, [45] |
| 8 | Az-H4 | 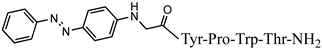 |
C43H47N9O7 | anticonvulsant activity, [46] |
| 9 | Rh-1 | rhodamineB-Gly-Tyr-Pro-Trp-Thr-NH2 | C59H69N9O9 | antiviral activity, [47] |
| 10 | Rh-2 | rhodamineB-β-Ala-Tyr-Pro-Trp- Thr-NH2 | C60H71N9O9 | antiviral activity, [47] |
| 11 | Rh-3 | rhodamineB-γ-Abu-Tyr-Pro-Trp-Thr-NH2 | C61H73N9O9 | antiviral activity, [47] |
| Hemorphin-5 analogs | ||||
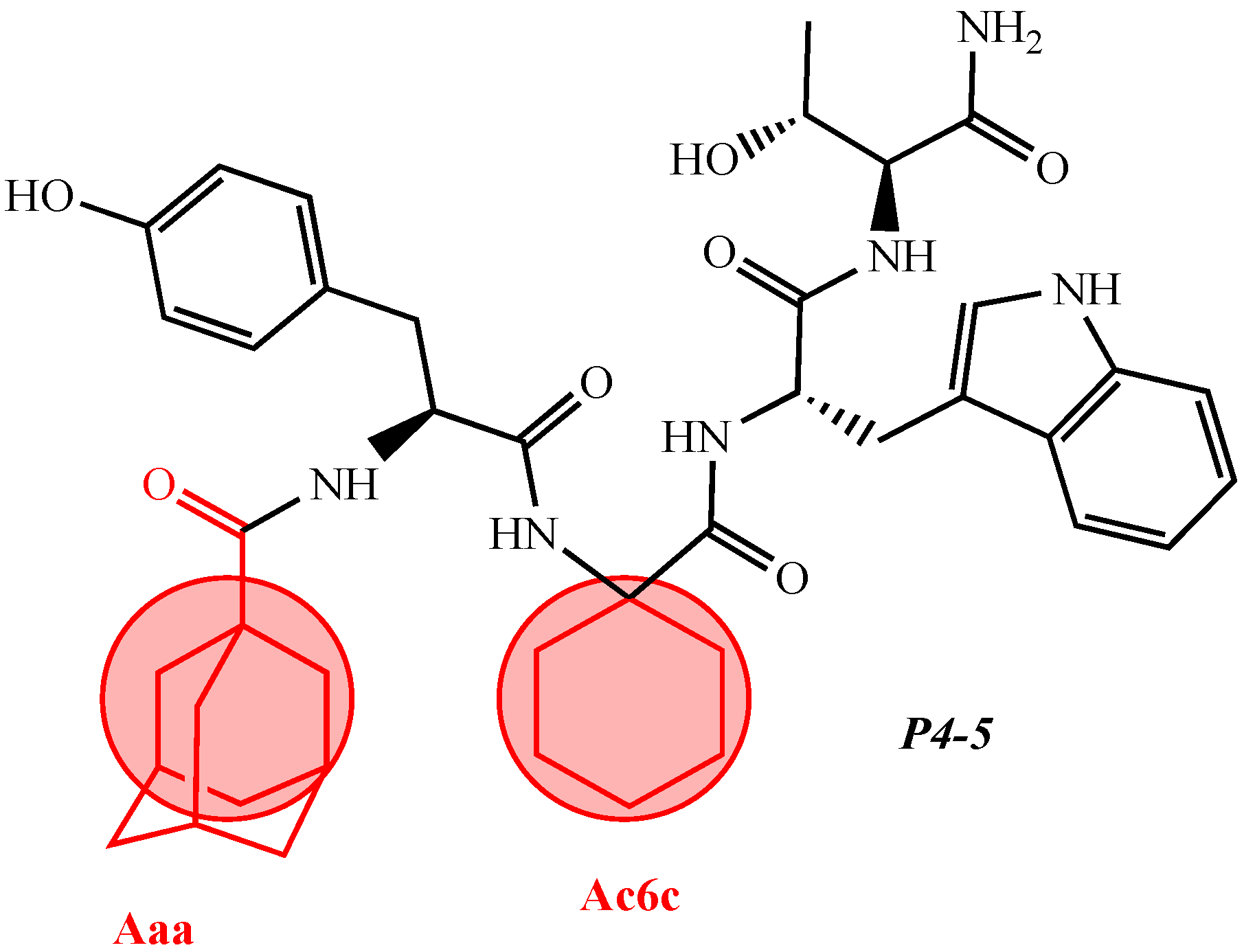
| 12 | V2/H2 | Val-Val-Tyr-Pro-Trp-Thr-Dap-NH2 | C42H60N10O9 | antinociceptive and anticonvulsant activity, [48][49] |
| 13 | V3/H3 | Val-Val-Tyr-Pro-Trp-Thr-Dab-NH2 | C43H62N10O9 | antinociceptive and anticonvulsant activity, [48][49] |
| 14 | V4/H4 | Val-Val-Tyr-Pro-Trp-Thr-Orn-NH2 | C44H64N10O9 | antinociceptive and anticonvulsant activity, [48][49] |
| 15 | V5/H5 | Val-Val-Tyr-Pro-Trp-Thr-Lys-NH2 | C45H66N10O9 | antinociceptive and anticonvulsant activity, [48][49] |
| 16 | V6/H6 | Ile-Val-Val-Tyr-Pro-Trp-Thr-Gln-NH2 | C50H73N11O11 | antinociceptive and anticonvulsant activity, [48][49] |
| 17 | V7/H7 | Aib-Val-Val-Tyr-Pro-Trp-Thr-Gln-NH2 | C48H69N11O11 | antinociceptive and anticonvulsant activity, [48][49] |
| 18 | V2p | 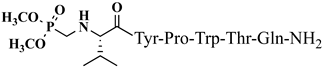 |
C42H60N9O12P | antinociceptive and anticonvulsant activity, [50][51] |
| 19 | V3p |  |
C43H62N9O12P | antinociceptive and anticonvulsant activity, [50][51] |
| 20 | V4p | 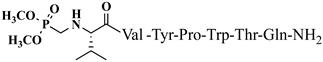 |
C47H69N10O13P | antinociceptive and anticonvulsant activity, [50][51] |
| 21 | V5p | 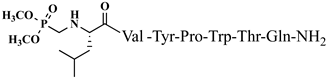 |
C48H71N10O13P | antinociceptive and anticonvulsant activity, [50][51] |
| 22 | V6p | 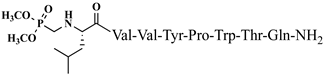 |
C53H80N11O14P | antinociceptive and anticonvulsant activity, [50][51] |
| 23 | Dm-5 | 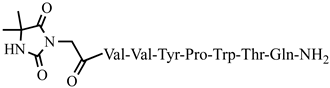 |
C51H70N12O13 | anticonvulsant activity, [45] |
| 24 | Ph-5 |  |
C61H74N12O13 | anticonvulsant activity, [45] |
| 25 | C-V | Cys-Val-Val-Tyr-Pro-Trp-Thr-Glu-NH2 | C47H66N10O12S | antiviral and antibacterial activity, [52] |
| 26 | H-V | His-Val-Val-Tyr-Pro-Trp-Thr-Glu-NH2 | C50H68N12O12 | antiviral and antibacterial activity, [52] |
| 27 | AC-V | Aaa-Cys-Val-Val-Tyr-Pro-Trp-Thr-Glu-NH2 | C58H80N10O13S | antiviral and antibacterial activity, [52] |
| 28 | AH-V | Aaa-His-Val-Val-Tyr-Pro-Trp-Thr-Glu-NH2 | C61H82N12O13 | antiviral and antibacterial activity, [52] |
| Hemorphin-7 analogs | ||||
| 29 | 2 | Val-Val-Tyr-Ac5c-Trp-Thr-Gln-Arg-Phe-NH2 | C60H85N15O12 | anticonvulsant activity, [53] |
| 30 | 3 | Val-Val-Tyr-Ac6c-Trp-Thr-Gln-Arg-Phe-NH2 | C61H87N15O12 | anticonvulsant activity, [53] |
| 31 | 4 | Val-Val-Tyr-Pro-Trp-Thr-Dap-Arg-Phe-NH2 | C57H81N15O11 | anticonvulsant activity, [53] |
| 32 | 5 | Val-Val-Tyr-Pro-Trp-Thr-Dab-Arg-Phe-NH2 | C58H83N15O11 | anticonvulsant activity, [53] |
| 33 | 6 | Val-Val-Tyr-Ac5c-Trp-Thr-Dap-Arg-Phe-NH2 | C58H83N15O11 | anticonvulsant activity, [53] |
| 34 | 7 | Val-Val-Tyr-Ac5c-Trp-Thr-Dab-Arg-Phe-NH2 | C59H85N15O11 | anticonvulsant activity, [53] |
| 35 | 8 | Val-Val-Tyr-Ac6c-Trp-Thr-Dap-Arg-Phe-NH2 | C59H85N15O11 | anticonvulsant activity, [53] |
| 36 | 9 | Val-Val-Tyr-Ac6c-Trp-Thr-Dab-Arg-Phe-NH2 | C60H87N15O11 | anticonvulsant activity, [53] |
| 37 | H7-1 | Ile-Val-Val-Tyr-Pro-Trp-Thr-Gln-Arg-D-Phe-NH2 | C65H94N16O13 | anticonvulsant activity, [54] |
| 38 | H7-2 | Ile-Val-Tyr-Pro-Trp-Thr-Gln-Arg-D-Phe-NH2 | C60H85N15O12 | anticonvulsant activity, [54] |
| 39 | H7-3 | D-Leu-Val-Val-Tyr-Pro-Trp-Thr-Gln-Arg-D-Phe-NH2 | C65H94N16O13 | anticonvulsant activity, [54] |
| 40 | H7-4 | D-Val-Val-Tyr-Pro-Trp-Thr-Gln-Arg-D-Phe-NH2 | C59H83N15O12 | anticonvulsant activity, [54] |
| 41 | H7-5 | 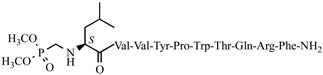 |
C68H101N16O16P | anticonvulsant activity, [54] |
| 42 | H7-6 | 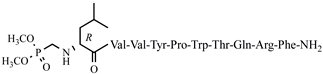 |
C68H101N16O16P | anticonvulsant activity, [54] |
| 43 | H7-7 |  |
C62H90N15O15P | anticonvulsant activity, [54] |
| 44 | H7-8 |  |
C62H90N15O15P | anticonvulsant activity, [54] |
| 45 | Dm-7 | 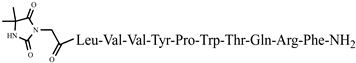 |
C72H102N18O16 | anticonvulsant activity, [45] |
| 46 | Ph-7 | 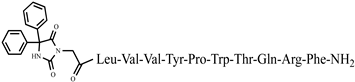 |
C82H106N18O16 | anticonvulsant activity, [45] |
| 47 | RGD1 | Val-Val-Tyr-Pro-Trp-Thr-Gln-Arg-Phe-Arg-Gly-Asp-NH2 | C71H103N21O17 | antinociceptive activity, [55] |
| 48 | RGD2 | Asp-Gly-Arg-Val-Val-Tyr-Pro-Trp-Thr-Gln-Arg-Phe-Arg-Gly-Asp-NH2 | C83H123N27O22 | antinociceptive activity, [55] |
| 49 | NH7C | Nic-Leu-Val-Val-Tyr-Pro-Trp-Thr-Glu-Arg-Phe-Cys-NH2 | C74H101N17O16S | antiviral and antibacterial activity, [52] |
| 50 | NCH7 | Nic-Cys-Leu-Val-Val-Tyr-Pro-Trp-Thr-Glu-Arg-Phe-NH2 | C74H101N17O16S | antiviral and antibacterial activity, [52] |
| Drug | TPE a | ED50 b µg | 95% Confidence Interval | TD50 c | PI d |
|---|---|---|---|---|---|
| (min) | |||||
| Phenytoin | 60 | 4.92 mg.kg−1 | (2.57–9.39) | >100 mg.kg−1 | >20.35 |
| Hemorphin-4 analogs | 10 | ||||
| P4-1 | - | - | - | - | |
| P4-2 | 2.33 | (1.13–4.83) | >10 | >4.29 | |
| P4-3 | 1.66 | (1.24–2.24) | >10 | >6.02 | |
| P4-4 | 2.33 | (1.13–4.83) | >10 | >4.29 | |
| P4-5 | 0.41 | (0.19–0.90) | >10 | >24.39 | |
| Peptide-based chemosensor bearing azobenzene side chain bio photoswitch | 10 | ||||
| Cis Az-H4 | 1.71 | (1.16–2.51) | >10 | >5.85 | |
| Trans A-H4 | 1.51 | (1.04–2.02) | >10 | >6.62 | |
| VV-Hemorphin-5 analogs | 10 | ||||
| V2 | - | - | - | - | |
| V3 | - | - | - | - | |
| V4 | 3.63 | (2.45–5.38) | >20 | >5.51 | |
| V5 | 3.19 | (2.62–3.87) | >20 | >6.27 | |
| V6 | 16.77 | (11.08–25.36) | >20 | >1.19 | |
| V7 | 16.55 | (12.78–21.41) | >20 | >1.21 | |
| 5,5-dimethyl- and 5,5-diphenylhydantoin-conjugated hemorphin derivatives | 10 | ||||
| Dm-4 | 0.36 | (0.13–1.0) | >3 | >8.33 | |
| Dm-5 | 0.74 | (0.06–8.8) | >5 | >6.76 | |
| Dm-7 | 0.7 | (0.05–9.58) | >10 | >14.29 | |
| Ph-4 | 0.56 | (0.06–5.34) | >8 | >14.29 | |
| Ph-5 | 0.25 | (0.10–0.60) | >5 | >20 | |
| LVV- and VV-hemorphin-7 analogs | 10 | ||||
| H7-1 | - | - | - | - | |
| H7-2 | 0.94 | (0.36–2.47) | >8 | >8.51 | |
| H7-3 | 0.68 | (0.19–2.51) | >8 | >11.76 | |
| H7-4 | 2.54 | (1.38–4.64) | >15 | >5.91 | |
| H7-5 | 1.53 | (0.60–3.88) | >3 | >1.96 | |
| H7-6 | 0.38 | (0.13–1.15) | >3 | >7.89 | |
| H7-7 | 1.58 | (0.68–3.70) | >5 | >3.16 | |
| H7-8 | 1.67 | (1.11–2.51) | >7 | >4.19 |
| Drug | TPE a | ED50 b | 95% Confidence Interval | TD50 c | PI d |
|---|---|---|---|---|---|
| (min) | µg | ||||
| Hemorphin-4 analogs | 10 | ||||
| P4-1 | 0.52 | (0.33–0.82) | >5 | >9.62 | |
| P4-2 | 2.16 | (1.87–2.49) | >5 | >2.31 | |
| P4-3 | 0.83 | (0.57–1.19) | >5 | >6.02 | |
| P4-4 | 0.44 | (0.25–0.78) | >5 | >11.36 | |
| P4-5 | 0.64 | (0.40–1.02) | >5 | >7.81 | |
| VV-Hemorphin-5 analogs | 10 | ||||
| V2 | 9.97 | (9.07−10.90) | >20 | 2 | |
| V4 | 5.09 | (4.31–6.02) | >20 | 3.93 | |
| V5 | 9.89 | (8.64–11.34) | >20 | 2.02 | |
| V6 | 5.55 | (5.51–5.58) | >20 | 7.84 | |
| V7 | 6.61 | (6.59–6.62) | >20 | 3.03 | |
| N-modified analogs of VV-hemorphin-5 with aminophosphonate moiety | 10 | ||||
| V2p | |||||
| V3p | 6.47 | (3.96–10.57) | >20 | 3.09 | |
| V4p | 4.31 | (2.76–10.47) | >20 | 4.64 | |
| V5p | 12.55 | (9.26–16.99) | >30 | 2.39 | |
| V6p | 14.11 | (9.17 –21.47) | >40 | 2.83 | |
| 5,5-dimethyl- and 5,5-diphenylhydantoin-conjugated hemorphin derivatives | 10 | ||||
| Dm-4 | 0.53 | (0.38–0.73) | >5 | 9.43 | |
| Dm-5 | 0.64 | (0.40–1.01) | >5 | 7.81 | |
| Dm-7 | 0.54 | (0.26–1.11) | >5 | 9.26 | |
| Ph-4 | 0.22 | (0.13–0.37) | >5 | 22.72 | |
| Ph-5 | 0.27 | (0.11–0.69) | >5 | 18.52 | |
| Ph-7 | 0.23 | (0.10–0.52) | >5 | 21.74 | |
| VV Hemorphin-7 analogs containing unnatural amino acids | 10 | ||||
| VV-H-2 | 5.69 | (3.67–8.81) | >30 | >5.27 | |
| VV-H-3 | 5.69 | (3.67–8.81) | >30 | >5.27 | |
| VV-H-4 | 3.83 | (1.50–9.76) | >20 | >5.22 | |
| VV-H-5 | 0.89 | (0.54–1.46) | >20 | >22.47 | |
| VV-H-6 | 2.67 | (4.67–8.19) | >20 | >7.49 | |
| VV-H-7 | 0.89 | (0.66–2.00) | >20 | >22.47 | |
| VV-H-8 | 1.01 | (0.29–3.55) | >20 | >19.80 | |
| VV-H-9 | 1.09 | (0.40–3.00) | >30 | >27.52 | |
| LVV- and VV-hemorphin-7 analogs | 10 | ||||
| H7-1 | 0.33 | (0.32–0.33) | >5 | >15.15 | |
| H7-2 | 2.6 | (1.58–4.55) | >5 | >1.87 | |
| H7-3 | - | - | - | - | |
| H7-4 | - | - | - | - | |
| H7-5 | 2.16 | (1.70–2.74) | >5 | >2.31 | |
| H7-6 | 2.44 | (1.41–4.21) | >5 | >2.05 | |
| H7-7 | 2.16 | (1.70–2.74) | >5 | >2.31 | |
| H7-8 | 3.03 | (2.44–3.74) | >5 | >1.65 |
| Drug | TPE a | Comparison of Activity Related to the Threshold Dose (µg/10 µL) for Clonic Seizures |
|---|---|---|
| (min) | ||
| Hemorphin-4 analogs | 10 | |
| P4 | ||
| P4-2 | P4-4 = P4-5 > P4-2 = P4-3 > P4 | |
| P4-3 | ||
| P4-4 | ||
| P4-5 | ||
| VV-Hemorphin-5 analogs | 10 | |
| V1 | ||
| V2 | V1 = V4 > V2 | |
| V4 | ||
| V5 | ||
| V6 | ||
| V7 | ||
| N-modified analogs of VV-hemorphin-5 with aminophosphonate moiety | 10 | V1 = V3p |
| V1 | ||
| V2p | ||
| V3p | ||
| V4p | ||
| V5p | ||
| V6p | ||
| Hemorphin-7 analogs containing unnatural amino acids | 10 | VV–H7 = V–H4 |
| VV-H7 | ||
| VV-H-2 | ||
| VV-H-3 | ||
| VV-H-4 | ||
| VV-H-5 | ||
| VV-H-6 | ||
| VV-H-7 | ||
| VV-H-8 | ||
| VV-H-9 | ||
| LVV- and VV-hemorphin-7 analogs | 10 | |
| H7 | H7-5> H7 = H7-3 = H7-6 = H7-7 = H7-8 > H7-1 | |
| H7-1 | ||
| H7-2 | ||
| H7-3 | ||
| H7-4 | ||
| H7-5 | ||
| H7-6 | ||
| H7-7 | ||
| H7-8 |
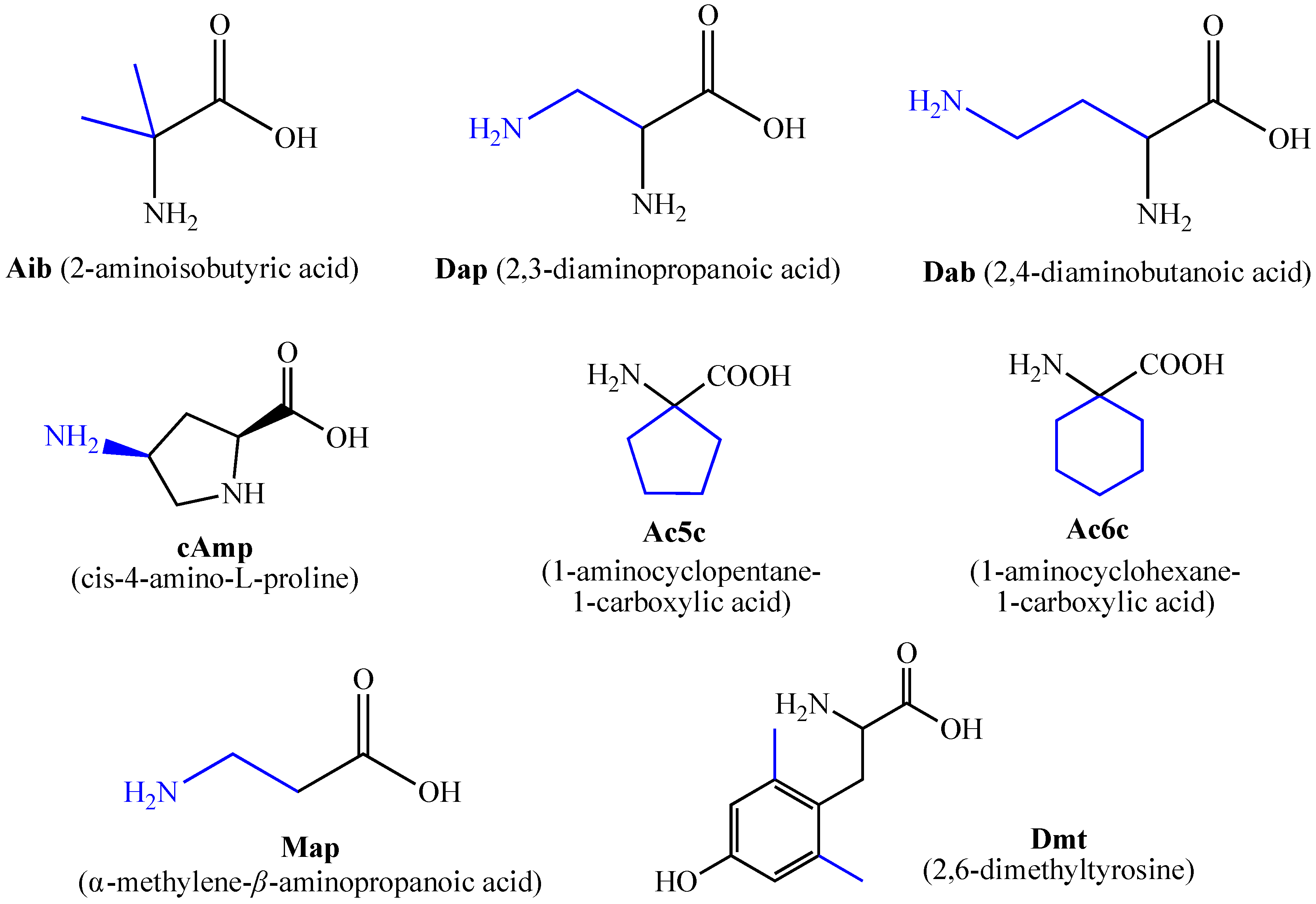
For the first time, α-aminophosphonic acids have been introduced into hemorphin peptides (compounds № 18–22 and 41–44) [50][51]. α-Aminophosphonates and aminophosphonic acids occupy an essential place among compounds containing a P-C bond and an amino group. They are structural analogs of natural α-amino acids, which are the “building blocks” of peptides and proteins. Their structure is of interest due to their diverse biological role. The obtained N-modified analogs of VV-hemorphin-5 containing an aminophosphonic residue have been described in detail in terms of structure-activity and have been investigated for antinociceptive and anticonvulsant activity. In the literature, it has been reported that the most potent hemorphin derivative was the V3p, with the lowest ED50 of 4.31 µg against psychomotor seizures and ivPTZ clonic seizures (Tables 3 and 4) [50]. The results of the docking study of the obtained in vivo results suggest that binding to the k-opioid receptor is the most likely mechanism of action of the peptide derivatives with anticonvulsant activity. These data lead to hypothesize that modification of the two N-terminal Val in the peptide molecules with an aminophosphonate residue in phosphopeptide analogs leads to significant changes in peptide activity and affinity [50][51].
For the first time, C-5-substituted hydantoins were introduced into hemorphins, aiming for a synergistic effect to enhance anticonvulsant activity (compounds № 6, 7, 23, 24, 45, and 46) [45]. Of these hybrid structures, the strongest anticonvulsant activity was reported for VV-hemorphin-5, possessing a 5,5′-diphenylhydantoin residue at the N-terminus and a hydrophobic Val–Val–Tyr–Pro–Trp–Thr–Gln–CONH2 amino acid sequence of the peptide molecule (Ph5). This compound showed low ED50 for MES and the 6 Hz test, respectively, compared to other tested peptide analogs (Tables 2 and 3). In silico analysis suggests that the underlying mechanism of the anticonvulsant effect of Ph-5 involves blocking sodium channels [45].
A series of Phe-modified analogs of hemorphin-7-NH2 were synthesized and characterized by replacing Phe at position 7 with various natural and unnatural amino acids: Leu, MePhe, D-Phe, Tic, Trp, Met, Oic, Phg (phenylglycine), pNO2Phe, Nle (norleucine), pClPhe, Thi, and Cha. Of all synthetic analogs, the most active are those containing unnatural amino acids: tetrahydro-isoquinoline-3-carboxylic acid (Tic), pClPhe, 3-thienylalanine (Thi), octahydroindole-2-carboxylic acid (Oic), and 3-cyclohexylalanine (Cha). Using phenytoin (5,5′-diphenylhydantoin) as a sodium channel blocker, it has been hypothesized that LVV-hemorphin-7 analogs activate the sympathetic nervous system via interaction with specific receptors functionally linked to phenytoin-sensitive sodium channels. Substitution of Arg at position 6 with Lys slightly reduced blood pressure, in contrast to its substitution with the amino acids citrulline, D-Arg, NO2Arg, Orn, or Ala, where it was significant [28]. Conversion of the C-terminal –COOH group with its amide –CONH2 in this type of compound significantly increased the activity of the corresponding peptide analog, indicating that the C-terminal –COOH group is not essential for activity. One possible reason for this is that such a change in the molecule leads to an increase in the resistance of the peptide to enzymatic degradation by endogenous carboxypeptidases [28][36][37]. Using proteomic studies, the biological role of LVV- and VV-hemorphin-7 as potential biomarkers in patients with posterior cranial fossa brain tumors has been demonstrated. It has been found that the presence of these two hemorphins can be used in the clinical diagnosis of this disease. In the presence of a brain tumor, both hemorphins are not detected in cerebrospinal fluid (CSF) analysis. At the same time, in the case of postoperative removal, they are present [72][73].
Two new N- and C-modified analogs of VV-hemorphin-7 containing RGD (Arg–Gly–Asp) residues as potential nociceptive agents and bioactive materials have been elucidated in detail № 47 and 48) [55]. From the eight LVV- and VV-hemorphin-7 analogs (compounds № 37–44), the H7-1 peptide analog showed the highest potency against the 6 Hz psychomotor seizures with ED50 of 0.33 µg (Table 3). However, while the H7-6 had the lowest ED50 in the MES test (Table 4), the H7-5 peptide analog raised the ivPTZ-induced clonic seizure at the highest rate at the doses used among the eight synthetized LVV- and VV-hemorphin-7 analogs (Table 4) [54]. Therefore, the modification at the N- and C-terminus with certain amino acids seems to play a critical role in the design of new LVV- and VVhemorphin-7 analogs.
Todorov et al. have synthesized and characterized a series of new analogs of VV-hemorphin-7 (compounds № 29-36) with potential anticonvulsant activity, modified with unnatural amino acids, following the structure Val-Val-Tyr-Xxx-Trp-Thr-Yyy-Arg-Phe-NH2, where Xxx is Ac5c (1-aminocyclopentane carboxylic acid) or Ac6c (1-aminocyclohexanecarboxylic acid) and Yyy is Dap (2,3-diaminopropane acid) or Dab (2,4-diaminobutanoic acid) [53]. The peptide analog VV-H5, containing diaminobutanoic acid in its molecule, showed the highest anticonvulsant activity. Moreover, this peptide analog had the lowest ED50 of 0.89 µg against psychomotor seizures and ED50 of 0.38 µg against the MES among the eight novel compounds (Tables 2 and 3). In addition, this peptide analog increased the threshold for ivPTZ clonic seizures at the lowest dose of 5 µg injected (Table 4). Interestingly, VV-H5 differs from VV-H4 by only one -CH2 group in the molecule, which is crucial for the anticonvulsant activity of this hemorphin derivative.
2.1. Analytical Characteristics of Hemorphin Analogs
It is known that some identical amino acid residues can have different reactivity with respect to given chemical reagents. For example, in an enzyme molecule, only one or a small number of side chains of amino acid residues located in the "active" center can bind substrates or coenzymes, while others with the same chemical composition cannot. As is known, a large part of the hydrophobic side chains are located in the interior of the molecule, thus building a compact core, while the polar and electron-charged groups are supported on the surface of this matrix. Moreover, the physical and chemical properties of the functional groups are strongly influenced by the nature of the microenvironment. Peptides exhibit partial solubility in aqueous (phosphate buffer, pH 6.86±0.01) and (organic) environments, with varying degrees of hydrophilicity and hydrophobicity (Figure 5). Ph-4 and Dm-4 show the greatest hydrophobicity, and Dm-5 and Ph-5 show the greatest tendency to dissolve in organic media. This is due to the fact that the attachment of a non-water-soluble hydantoin component to the main short-chain peptide scaffold stabilizes the zwitterionic form in solutions with a pH of about 7 and interferes with solubility in aqueous solutions. For these compounds, the isoelectric points are around 7.0. рI values close to and around 7 are observed for most short-chain peptide modifications (Figure 6). Compounds P4-4 and P4-5 with modifications Ac5c, Ac6c have pI values around 7, and it is the zwitterionic form in which they will be at this pH that will interfere with their solubility when preparing, for example, injection solutions for biological analyses. Peptide forms with these modifications also showed the least pronounced biological activities. As an important parameter evaluating the behavior of non-peptides in solution are also acid-base constants. Determination of the equilibrium constants (pK) of proton dissociation from ionizable side chains of amino acids represents a very important application of spectroscopy and electrochemistry in peptide chemistry. This definition allows conclusions to be drawn regarding the location of these groups in the peptide matrix, as well as their involvement in various interactions. As mentioned the amino acid fragment of the hemorphin molecule: Tyr-Pro-Trp is the main sequence thanks to which receptor binding takes place. On the other hand, the amino acids tyrosine and tryptophan, bonded in a peptide chain, are one of the main amino acids exhibiting fluorescent, electrochemical and acid-basic properties. Table 5 gives the determined pK values of the hemorphin peptides, calculated by applying different analytical techniques, most often by potentiometric titration or mathematical processing of data from the fluorescence/voltammetric analysis. Hemorphine derivatives have acidic properties, which turn them into protolytes of different strengths depending on the amino acid radicals: the more acidic amino acids in the peptide, the stronger its acidic properties are expressed. Regardless of the peptide modifications made, the determined acidity constants refer to the side P-groups of tyrosine, the indole nucleus of tryptophan, and arginine in the arginine-containing peptides, exhibiting different degrees of polarity at a pH close to the physiological values of 6-8 (corresponding to the conditions of the cell cytosol). Most peptide derivatives showed approximate pK values related to proton exchange with the -OH group of tyrosine and the indole moiety of tryptophan. As can be seen, peptide derivatives containing a phosphonic group adjacent to the amino acid tyrosine (V2P-V3P series) have weaker protolytic properties, and the protolytic power of long-chain hemorphin derivatives increases with the distance of the -OH group of tyrosine from the corresponding structural modification.
Table 5. Values of acid-base constants (pK) of some peptides.
|
Peptide |
pKa1; pKa2 Constants |
|
|
|
|
|
|
P4 |
by potentiometric titration |
3.80; 6.44, [44] |
|
P4-1 |
3.89; 6.52, [44] |
|
|
P4-2 |
3.93; 6.71, [44] |
|
|
P4-3 |
3.88; 6.93, [44] |
|
|
P4-4 |
6.16; 8.90, [44] |
|
|
P4-5 |
6.20; 9.06, [44] |
|
|
Dm-4 |
by potentiometric titration |
2.86; [44] |
|
Ph-4 |
by potentiometric titration |
2.98; [45] |
|
Rh-1 |
by potentiometric titration |
2.81; 6.60, [47] |
|
Rh-2 |
2.78;6.38, [47] |
|
|
Rh-3 |
2.86;6.39, [47] |
|
|
Hemorphin-5 analogs |
||
|
V2/H2 |
by potentiometric titration |
|
|
V3/H3 |
||
|
V4/H4 |
||
|
V5/H5 |
||
|
V6/H6 |
||
|
V7/H7 |
||
|
V2p |
by potentiometric titration and voltamperometry |
|
|
V3p |
||
|
V4p |
||
|
V5p |
||
|
V6p |
||
|
Dm-5 |
by potentiometric titration |
3.06; 7.14, [45] |
|
Ph-5 |
3.09; 6.98, [45] |
|
|
C-V |
by fluorimetry |
5.18, [52] |
|
H-V |
4.75, [52] |
|
|
AC-V |
5.43, [52] |
|
|
AH-V |
4.84, [52] |
|
|
Hemorphin-7 analogs |
||
|
2 |
by potentiometric titration |
8.04(Val); 5.34(Tyr), [53] |
|
3 |
7.49(Val); 4.83(Tyr), [53] |
|
|
4 |
7.10(Val);5.46(Dap, Dab); 3.14(Tyr), [53] |
|
|
5 |
8.08(Val);7.21(Dap, Dab); 5.98(Tyr),[53] |
|
|
6 |
8.15(Val);6.87(Dap, Dab); 4.72(Tyr), [53] |
|
|
7 |
8.21(Val);7.26(Dap, Dab); 4.70(Tyr), [53] |
|
|
8 |
9.20(Val);8.03(Dap, Dab); 5.27(Tyr), [53] |
|
|
9 |
9.08(Val);8.80(Dap, Dab); 4.66(Tyr), [53] |
|
|
H7-1 |
by potentiometric titration |
2.98; 6.12, [54] |
|
H7-2 |
3.09; 6.62, [54] |
|
|
H7-3 |
3.05; 6.78, [54] |
|
|
H7-4 |
3.22; 6.52, [54] |
|
|
H7-5 |
3.17; 6.23, [54] |
|
|
H7-6 |
2.98; 5.85, [54] |
|
|
H7-7 |
3.15; 6.09, [54] |
|
|
H7-8 |
2.78; 5.52, [54] |
|
|
Dm-7 |
by potentiometric titration |
3.19; 5.11, [45] |
|
Ph-7 |
3.23; 6.45, [45] |
|
|
RGD1 |
by potentiometric titration |
3.53; 6.42, [55] |
|
RGD2 |
3.48; 6.34, [55] |
|
|
NH7C |
by fluorimetry |
5.07, [52] |
|
NCH7 |
4.78, [52] |
|
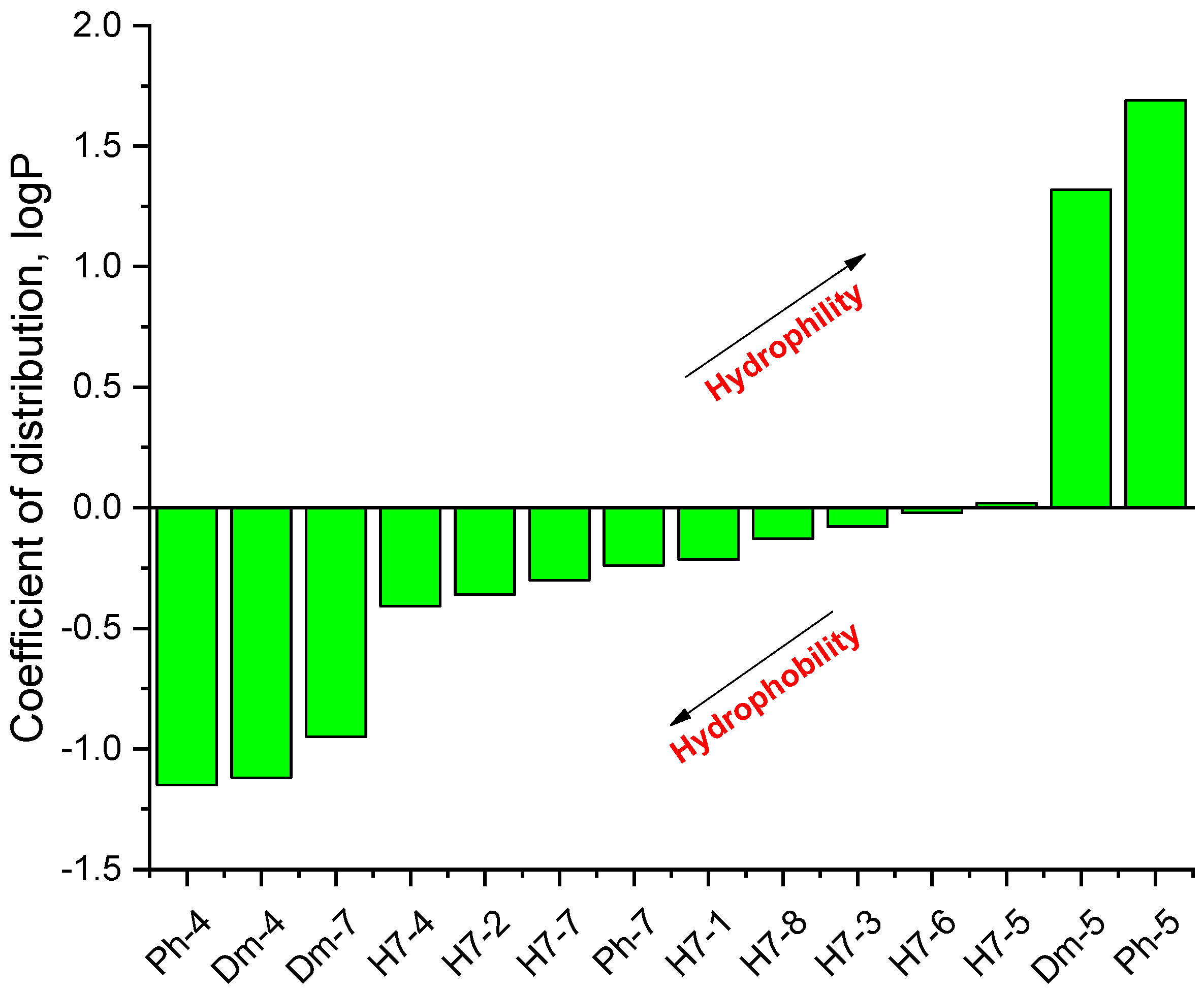
Figure 5. Logarithmic values of the partition coefficient (log P) of some of the investigated hemorphin compounds.
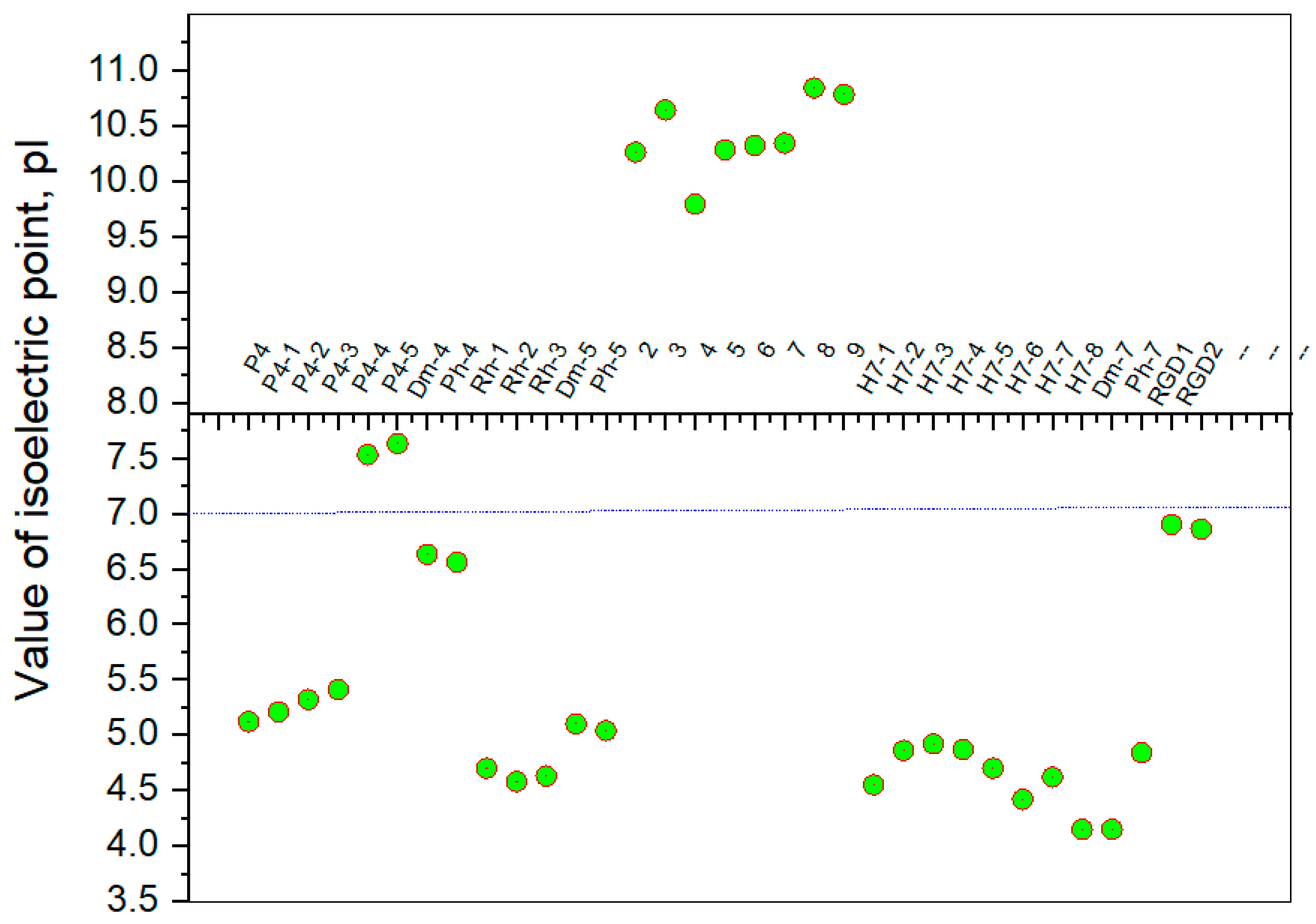
Figure 6. Summary plot of isoelectric point (pI) values of hemorphin analogs.
2.2. Electrochemical Behavior of Hemorphin Analogs
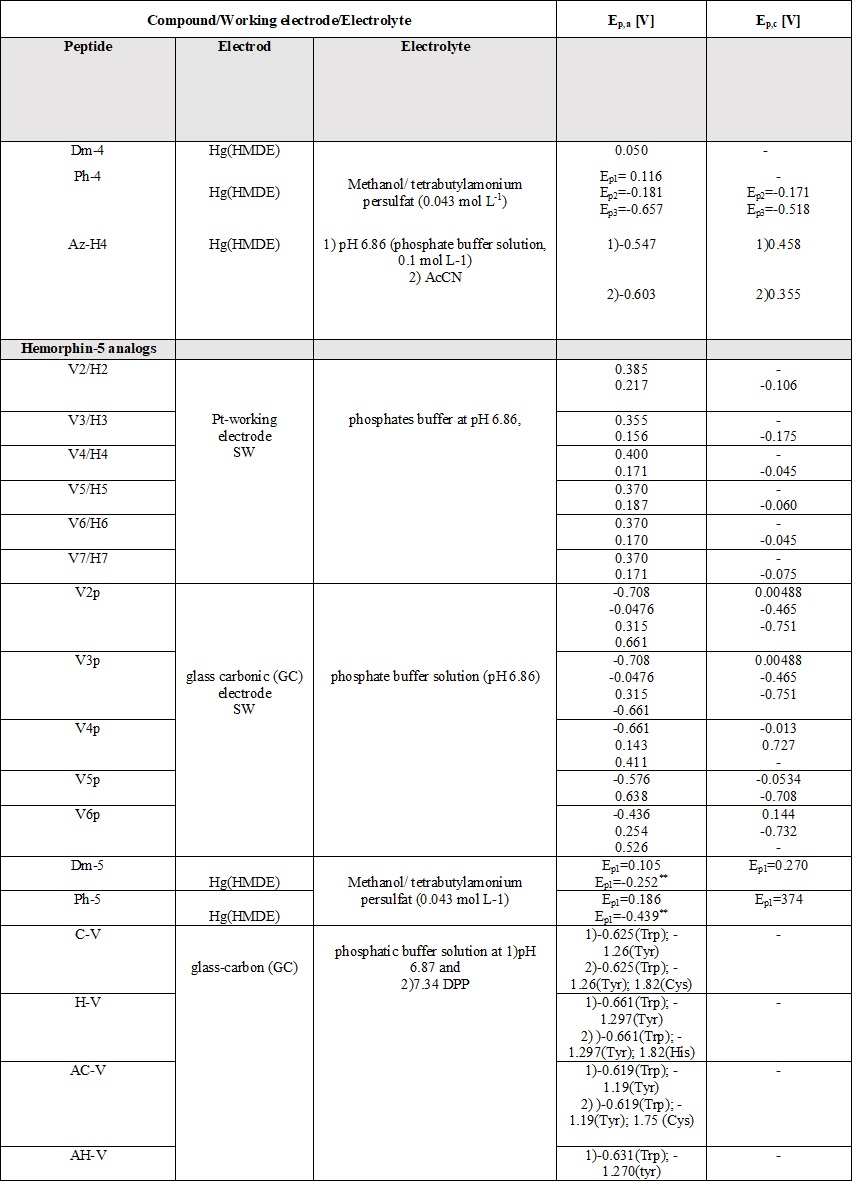
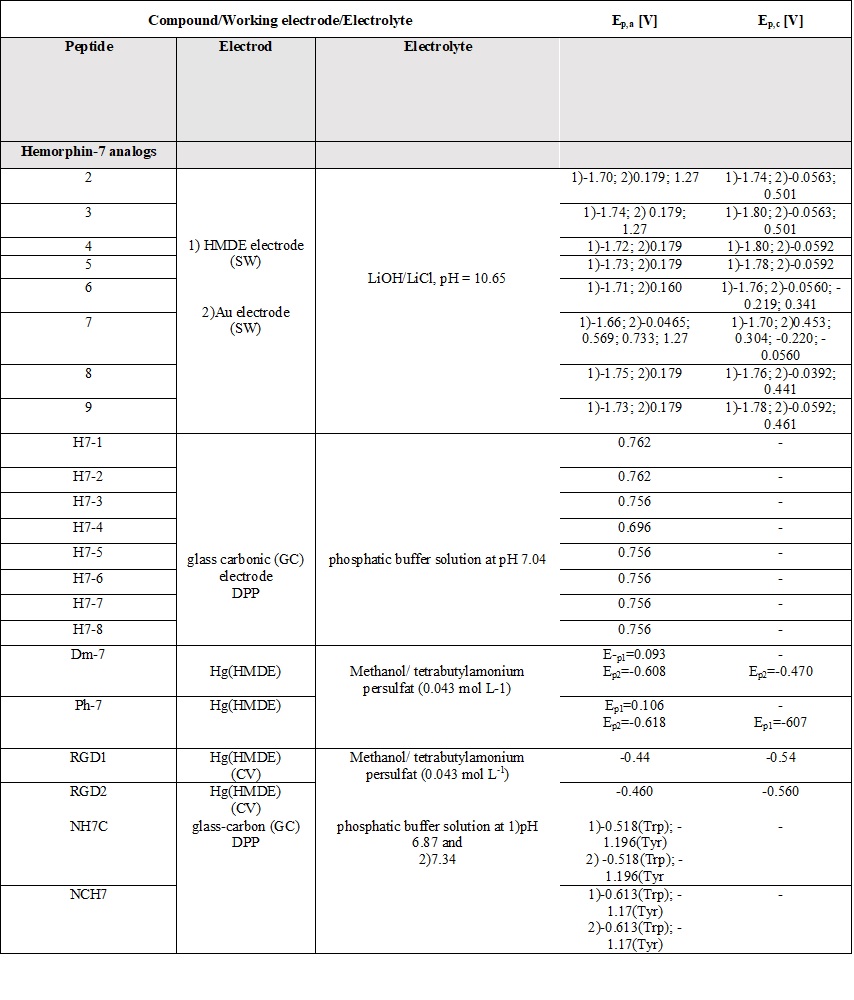
A comparative review of the electrochemical properties of peptide derivatives would be of interest from a scientific point of view, as the method offers the elucidation of basic mechanisms of action and the detection of molecular and structural modifications (aggregation). Electrochemical methods add a fresh, new perspective to the research field of hemorphin analogs in terms of their rapid detection, characterization, the study of redox behavior, and electrode response nature. Most of the studied hemorphin derivatives contain only the voltammetrically active tyrosine and tryptophan, connected in the sequence -Tyr-Pro-Trp- and their oxidation occurs thanks to the presence of p-electrons at the hydroxyl group of the phenol part (Tyr) and indole ring (Trp), respectively [48][49][50]. Studies were carried out in different electrolyte environments by using differently charged surfaces (Table 6). On a glass carbon electrode, at pH⁓7, the oxidation of hemorphin analogs is irreversible, which occurs at positive potentials (Ep⁓+0.4V for Vp series and Ep⁓+0.7 for H7 series V vs. Ag/AgCl), corresponding to the signal of tyrosine due to –OH group of the phenol part which is oxidized to a glass carbonic electrode at potential closed to +0.7[74]. The oxidation of histidine and cysteine to peptides containing them at a glassy carbon electrode is an irreversible, diffusion-controlled, and pH-dependent process (Table 6) that occurs at Ep ⁓ 1.2 V, vs. Ag/AgCl[75]. A medium of tetrabutylammonium persulfate with methanol proved to be favorable for the detection and determination of hemorphin analogs at mercury electrodes, with the resulting well-shaped reduction/oxidation peaks indicating reversible to quasi-reversible electrode processes. Analyzes showed that the reactivity of the tyrosine and tryptophan regions was conserved. This, together with the fact that the concentration dependence of the current signal is proportional, gives the reason to conclude that in these environments the structure of the compounds is preserved and aggregation does not occur regardless of the modification of the molecule. Electrochemical methods of investigating the aggregation and fibrillization of hemorphin peptides could become important additional tools in biochemical research, leading to new insights into the molecular mechanisms underlying pathogenesis.
Table 6. Parameters of the voltammetric measurements and the electrochemical data of peptides [45][46][48][49][50][51][52][53][54][55].
3. Conclusions
Researchers showed that more than 50 new hemorphin analogs obtained by solid-phase peptide synthesis by the Fmoc-strategy have been synthesized and characterized up to date. These hemorphin peptides contain various unnatural and unusual amino acids, D-amino acids, α-aminophosphonic acids. Structure-activity relationship studies show that not only the position of the modification, but also the nature of the amino acid involved leads to significant changes in the physicochemical properties (change in pKa, pI, and logP of the peptide), biological activity, and receptor affinity. It can be summarized that even the smallest change in the hemorphin molecule has a great influence on the biological activity, such as the introduction of the unnatural amino acid Dap, and the replacement of Pro with the conformationally constrained amino acids (Ac5c, Ac6c, and adamantane moieties). Structure-activity analysis revealed that the incorporation of an adamantane residue at the N-terminus is necessary for protection against the spread of seizures. Data obtained so far have shown that modification of the two N-terminal Vals in peptide molecules by an aminophosphonate residue in phosphopeptide analogs requires increased biological activity and receptor affinity, from which it can be concluded that the successful design of new analogs of LVV- and VV-hemorphin-7 involves modification of the N- and C-termini with specific amino acids. Predictions from the docking analysis suggest that binding to the k-opioid receptor is the most relevant mechanism of action for the new peptide analogs that possess unnatural amino acids. All hemorphin analogs can be successfully detected at low concentrations by applying voltammetric techniques on solid and mercury electrodes. Electrochemical methods are a good alternative for proving molecular conformational changes and processes of aggregation and peptide fibrillization and can be applied in studying the properties of hemorphin peptides in environments with different matrix compositions.
References
- Mielczarek, P.; Hartman, K.; Drabik, A.; Hung, H.-Y.; Huang, E.Y.-K.; Gibula-Tarlowska, E.; Kotlinska, J.H.; Silberring, J. Hemorphins—From Discovery to Functions and Pharmacology. Molecules 2021, 26, 3879.
- Fukui, K.; Shiomi, H.; Takagi, H.; Hayashi, K.; Kiso, Y.; Kitagawa, K. Isolation from bovine brain of a novel analgesic pentapeptide, neo-kyotorphin, containing the Tyr-Arg (kyotorphin)unit. Neuropharmacology 1983, 22, 191–196.
- Schechter, A.N. Hemoglobin research and the origins of molecular medicine. Blood J. Am. Soc. Hematol. 2008, 112, 3927–3938.
- Liu, L.; Zeng, M.; Stamler, J.S. Hemoglobin induction in mouse macrophages. Proc. Natl. Acad. Sci. USA 1999, 96, 6643–6647.
- Newton, D.A.; Rao, K.M.K.; Dluhy, R.A.; Baatz, J.E. Hemoglobin is expressed by alveolar epithelial cells. J. Biol. Chem. 2006, 281, 5668–5676.
- Wride, M.A.; Mansergh, F.C.; Adams, S.; Everitt, R.; Minnema, S.E.; Rancourt, D.E.; Evans, M.J. Expression profiling and gene discovery in the mouse lens. Mol. Vis. 2003, 9, 360–396.
- Setton-Avruj, C.P.; Musolino, P.L.; Salis, C.; Allo, M.; Bizzozero, O.; Villar, M.J.; Pasquini, J.M. Presence of α-globin mRNA and migration of bone marrow cells after sciatic nerve injury suggests their participation in the degeneration/regeneration process. Exp. Neurol. 2007, 203, 568–578.
- Ohyagi, Y.; Yamada, T.; Goto, I. Hemoglobin as a novel protein developmentally regulated in neurons. Brain Res. 1994, 635, 323–327.
- Schelshorn, D.W.; Schneider, A.; Kuschinsky, W.; Weber, D.; Krüger, C.; Dittgen, T.; Maurer, M.H. Expression of hemoglobin in rodent neurons. J. Cereb. Blood Flow Metab. 2009, 29, 585–595.
- Richter, F.; Meurers, B.H.; Zhu, C.; Medvedeva, V.P.; Chesselet, M.F. Neurons express hemoglobin α-and β-chains in rat and human brains. J. Comp. Neurol. 2009, 515, 538–547.
- Ivanov, V.T.; Karelin, A.A.; Philippova, M.M.; Nazimov, I.V.; Pletnev, V.Z. Hemoglobin as a source of endogenous bioactive peptides: The concept of tissue-specific peptide pool. Pept. Sci. 1997, 43, 171–188.
- Karelin, A.A.; Blishchenko, E.Y.; Ivanov, V.T. Fragments of Functional Proteins: Role in Endocrine Regulation. Neurochem. Res. 1999, 24, 1117–1124.
- Giardina, B. Hemoglobin: Multiple molecular interactions and multiple functions. An example of energy optimization and global molecular organization. Mol. Asp. Med. 2022, 84, 101040.
- Amanat, A.; Alzeyoudi, C.A.; Almutawa, A.C.; Alnajjar, A.H.; Vijayan, R. Molecular basis of the therapeutic properties of hemorphins, Pharmacol. Res. 2020, 158, 104855.
- Yoshikawa, M. Bioactive peptides derived from natural proteins with respect to diversity of their receptors and physiological effects. Peptides 2015, 72, 208–225.
- Brantl, V.; Gramsch, C.; Lottspeich, F.; Mertz, R.; Jaeger, K.H.; Herz, A. Novel opioid peptides derived from hemoglobin: Hemorphins. Eur. J. Pharmacol. 1986, 125, 309–310.
- Yang, Y.R.; Chiu, T.H.; Chen, C.L. Structure–activity relationships of naturally occurring and synthetic opioid tetrapeptides acting on locus coeruleus neurons. Eur. J. Pharmacol. 1999, 372, 229–236.
- Mollica, A.; Pinnen, F.; Stefanucci, A.; Mannina, L.; Sobolev, A.P.; Lucente, G.; Davis, P.; Lai, J.; Ma, S.-W.; Porreca, F.; et al. cis-4-Amino-l-proline Residue as a Scaffold for the Synthesis of Cyclic and Linear Endomorphin-2 Analogues: Part 2. J. Med. Chem. 2012, 55, 19–8477.
- Mollica, A.; Stefanucci, A.; Costante, R.; Novellino, E. Pyroglutamic Acid Derivatives: Building Blocks for Drug Discovery. HETEROCYCLES 2014, 89, 1801.
- Mollica, A.; Stefanucci, A.; Costante, R.; Hruby, V.J. Chapter 2—Rational Approach to the Design of Bioactive Peptidomimetics: Recent Developments in Opioid Agonist Peptides. Stud. Nat. Prod. Chem. 2015, 46, 27–68.
- Schiller, P.W. Development of opioid peptide analogs as pharmacologic tools and as potential drugs: Current status and future directions. NIDA Res. Monogr. 1991, 112, 180–197.
- Blishchenko, E.Y.; Sazonova, O.V.; Kalinina, O.A.; Yatskin, O.N.; Philippova, M.M.; Surovoy, A.Y.; Karelin, A.A.; Ivanov, V.T. Family of hemorphins: Co-relations between amino acid sequences and effects in cell cultures. Peptides 2002, 23, 903–910.
- Maurer, R.; Römer, D.; Büscher, H.H.; Gähwiler, B.H.; Thies, P.W.; David, S. Valorphin: A novel chemical structure with opioid activity. Neuropeptides 1985, 5, 387–390.
- Erchegyi, J.; Kastin, A.J.; Zadina, J.E.; Qiu, X.D. Isolation of a heptapeptide Val-Val-Tyr-Pro- Trp-Thr-Gln (valorphin) with some opiate activity. Int. J. Pept. Protein Res. 1992, 39, 477–484.
- Blishchenko, E.; Sazonova, O.; Surovoy, A.; Khaidukov, S.; Sheikine, Y.; Sokolov, D.; Ivanov, V. Antiproliferative action of valorphin in cell cultures. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2002, 8, 438–452.
- Blishchenko, E.Y.; Mernenko, O.A.; Mirkina, I.I.; Satpaev, D.K.; Ivanov, V.S.; Tchikin, L.D.; Ivanov, V.T. Tumor cell cytolysis mediated by valorphin, an opioid-like fragment of hemoglobin β-chain. Peptides 1997, 18, 79–85.
- Song, C.; Rahim, R.T.; Davey, P.C.; Bednar, F.; Bardi, G.; Zhang, L.; Zhang, N.; Oppenheim, J.J.; Rogers, T.J. PKC mediates-opioid receptor-induced cross-desensitization of chemokine receptor CCR5. J. Biol. Chem. 2011, 286, 20354–20365.
- Blishchenko, E.Y.; Sazonova, O.V.; Kalinina, O.A.; Moiseeva, E.V.; Vass, A.A.; Karelin, A.A.; Ivanov, V.T. Antitumor effect of valorphin in vitro and in vivo: Combined action with cytostatic drugs. Cancer Biol. Ther. 2005, 4, 125–131.
- Karelin, A.A.; Philippova, M.M.; Karelina, E.V.; Ivanov, V.T. Isolation of endogenous hemorphin-related hemoglobin fragments from bovine brain. Biochem. Biophys. Res. Commun. 1994, 202, 410–415.
- Moeller, I.; Lew, R.A.; Mendelsohn, F.A.; Smith, A.I.; Brennan, M.E.; Tetaz, T.J.; Chai, S.Y. The globin fragment LVV-hemorphin-7 is an endogenous ligand for the AT4 receptor in the brain. J. Neurochem. 1997, 68, 2530–2537.
- Murillo, L.; Piot, J.M.; Coitoux, C.; Fruitier-Arnaudin, I. Brain processing of hemorphin-7 peptides in various subcellular fractions from rats. Peptides 2006, 27, 3331–3340.
- Moisan, S.; Harvey, N.; Beaudry, G.; Forzani, P.; Burhop, K.E.; Drapeau, G.; Rioux, F. Structural requirements and mechanism of the pressor activity of Leu-Val-Val-hemorphin-7, a fragment of hemoglobin β-chain in rats. Peptides 1998, 19, 119–131.
- Wei, F.; Zhao, L.; Jing, Y. Hemoglobin-derived peptides and mood regulation. Peptides 2020, 127, 170268.
- Hung, H.Y.; Chow, L.H.; Kotlinska, J.H.; Drabik, A.; Silberring, J.; Chen, Y.H.; Huang, E.Y. LVV-hemorphin-7 (LVV-H7) plays a role in antinociception in a rat model of alcohol-induced pain disorders. Peptides 2021, 136, 170455.
- Ali, A.; Baby, B.; Soman, S.S.; Vijayan, R. Molecular insights into the interaction of hemorphin and its targets. Sci. Rep. 2019, 9, 1–16.
- Ali, A.; Alzeyoudi, S.A.R.; Almutawa, S.A.; Alnajjar, A.N.; Al Dhaheri, Y.; Vijayan, R. Camel hemorphins exhibit a more potent angiotensin-I converting enzyme inhibitory activity than other mammalian hemorphins: An in silico and in vitro study. Biomolecules 2020, 10, 486.
- Caballero, J. Considerations for docking of selective angiotensin-converting enzyme inhibitors. Molecules 2020, 25, 295.
- Amanat, A.; Soman, S.S.; Vijayan, R. Dynamics of camel and human hemoglobin revealed by molecular simulations. Sci. Rep. 2022, 12, 122.
- Ojima, I.; Lin, S.; Wang, T. Recent advances in the medicinal chemistry of taxoids with novel beta-amino acid side chains. Curr. Med. Chem. 1999, 6, 927–954.
- Mortensen, U.H.; Raaschou-Nielsen, M.; Breddam, K. Recognition of C-terminal amide groups by (serine) carboxypeptidase Y investigated by site-directed mutagenesis. J. Biol. Chem. 1994, 269, 15528–15532.
- Pogozheva, I.D.; Przydzial, M.J.; Mosberg, H.I. Homology modeling of opioid receptor-ligand complexes using experimental constraints. AAPS J. 2005, 7, E434–E448.
- Gademann, K.; Hintermann, T.; Schreiber, J.V. Beta-peptides: Twisting and turning. Curr. Med. Chem. 1999, 6, 905–925.
- Fülöp, F. The chemistry of 2-aminocycloalkanecarboxylic acids. Chem. Rev. 2001, 101, 2181–2204.
- Todorov, P.; Peneva, P.; Tchekalarova, J.; Georgieva, S.; Rangelov, M.; Todorova, N. Structure–activity relationship study on new Hemorphin-4 analogues containing steric restricted amino acids moiety for evaluation of their anticonvulsant activity. Amino Acids 2020, 52, 375–1390.
- Todorov, P.; Peneva, P.; Georgieva, S.; Tchekalarova, J.; Rangelov, M.; Todorova, N. Synthesis and characterization of new 5,5’-dimethyl- and 5,5’-diphenylhydantoin-conjugated hemorphin derivatives designed as potential anticonvulsant agents. New J. Chem. 2022, 46, 2198–2217.
- Todorov, P.; Georgieva, S.; Peneva, P.; Tchekalarova, J. Spectral and electrochemical solvatochromic investigations of newly synthesized peptide-based chemosensor bearing azobenzene side chain bio photoswitch. Dye. Pigment. 2021, 91, 109348.
- Todorov, P.; Georgieva, S.; Staneva, D.; Peneva, P.; Grozdanov, P.; Nikolova, I.; Grabchev, I. Synthesis of new modified with Rhodamine B peptides for antiviral protection of textile materials. Molecules 2021, 26, 6608.
- Todorov, P.; Peneva, P.; Pechlivanova, D.; Georgieva, S.; Dzhambazova, E. Synthesis, characterization and nociceptive screening of new VV-hemorphin-5 analogues. Bioorganic Med. Chem. Lett. 2018, 28, 3073–3079.
- Todorov, P.; Rangelov, M.; Peneva, P.; Todorova, N.; Tchekalarova, J. Anticonvulsant evaluation and docking analysis of VV-Hemorphin-5 analogues. Drug Dev. Res. 2019, 80, 425–437.
- Todorov, P.; Peneva, P.; Tchekalarova, J.; Rangelov, M.; Georgieva, S.; Todorova, N. Synthesis, characterization and anticonvulsant activity of new series of N-modified analogues of VV-Hemorphin-5 with aminophosphonate moiety. Amino Acids 2019, 51, 10–12.
- Assenov, B.; Pechlivanova, D.; Dzhambazova, E.; Peneva, P.; Todorov, P. Antinociceptive Effects of VV-Hemorphin-5 Peptide Analogues Containing Aminophosphonate Moiety in Mouse Formalin Model of Pain. Protein Pept. Lett. 2021, 28, 442–449.
- Todorov, P.; Georgieva, S.; Staneva, D.; Peneva, P.; Grozdanov, P.; Nikolova, I.; Vasileva-Tonkova, E.; Grabchev, I. Study of Novel Peptides for Antimicrobial Protection in Solution and on Cotton Fabric. Molecules 2022, 27, 4770.
- Todorov, P.; Peneva, P.; Tchekalarova, J.; Georgieva, S. Potential anticonvulsant activity of novel VV-hemorphin-7 analogues containing unnatural amino acids: Synthesis and characterization. Amino Acids 2020, 52, 567–585.
- Todorov, P.; Georgieva, S.; Peneva, P.; Tchekalarova, J. Investigation of the structure–activity relationship in a series of new LVV-and VV-hemorphin-7 analogues designed as potential anticonvulsant agents. Amino Acids 2022, 54, 261–275.
- Georgieva, S.; Todorov, P.; Nikolov, S.; Dzhambazova, E.; Peneva, P.; Assenov, B.; Pechlivanova, D. New N-and C-modified RGD-hemorphins as potential biomedical application on Ti-surface materials: Synthesis, characterization and antinociceptive activity. Mol. Divers. 2022, 1–18.
- De Rosa, T.F. Maximal Electroshock Seizure (MESs) are a model of generalized tonic-clonic seizures. From: Models of Seizures and Epilepsy, 2006. In Significant Pharmaceuticals Reported in US Patents, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2007; ISBN 978-0-08-045344-6.
- Potschka, H. Animal models of drug-resistant epilepsy. Epileptic Disord. 2012, 14, 226–234.
- Gredičak, M.; Supek, F.; Kralj, M.; Majer, Z.; Hollosi, M.; Smuc, T.; Mlinaric-Majerski, K.; Horvat, Š. Computational structure–activity study directs synthesis of novel antitumor enkephalin analogs. Amino Acids 2010, 38, 1185–1191.
- Löscher, W.; Klotz, U.; Zimprich, F.; Schmidt, D. The clinical impact of pharmacogenetics on the treatment of epilepsy. Epilepsia 2009, 50, 1–23.
- Alachkar, A.; Ojha, S.K.; Sadeq, A.; Adem, A.; Frank, A.; Stark, H.; Sadek, B. Experimental Models for the Discovery of Novel Anticonvulsant Drugs: Focus on Pentylenetetrazole-Induced Seizures and Associated Memory Deficits. Current Pharm. Design 2020, 26, 1693–1711.
- Prasad, S.; Rao, R.B.; Balaram, P. Contrasting solution conformations of peptides containing α, α-dialkylated residues with linear and cyclic side chains. Biopolym. Orig. Res. Biomol. 1995, 35, 11–20.
- Liu, J.; Obando, D.; Liao, V.; Lifa, T.; Codd, R. The many faces of the adamantyl group in drug design. Eur. J. Med. Chem. 2011, 46, 1949–1963.
- Wanka, L.; Iqbal, K.; Schreiner, P.R. The lipophilic bullet hits the targets: Medicinal chemistry of adamantane derivatives. Chem. Rev. 2013, 113, 3516–3604.
- Field, M.J.; Li, Z.; Schwarz, J.B. Ca2+ channel α2-δ ligands for the treatment of neuropathic pain. J. Med. Chem. 2007, 50, 2569–2575.
- Bryans, J.S.; Davies, N.; Gee, N.S.; Dissanayake, V.U.; Ratcliffe, G.S.; Horwell, D.C.; O’Neill, J.A. Identification of novel ligands for the gabapentin binding site on the α2δ subunit of a calcium channel and their evaluation as anticonvulsant agents. J. Med. Chem. 1998, 41, 1838–1845.
- Mart, R.J.; Allemann, R.K. Azobenzene photocontrol of peptides and proteins. Chem. Commun. 2016, 52, 12262–12277.
- Szymanski, W.; Beierle, J.M.; Kistemaker, H.A.; Velema, W.A.; Feringa, B.L. Reversible photocontrol of biological systems by the incorporation of molecular photoswitches. Chem. Rev. 2013, 113, 6114–6178.
- Piotto, S.; Trapani, A.; Bianchino, E.; Ibarguren, M.; López, D.J.; Busquets, X.; Concilio, S. The effect of hydroxylated fatty acid-containing phospholipids in the remodeling of lipid membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 1509–1517.
- Mendive-Tapia, L.; Preciado, S.; García, J.; Ramón, R.; Kielland, N.; Albericio, F.; Lavilla, R. New peptide architectures through C–H activation stapling between tryptophan–phenylalanine/tyrosine residues. Nat. Commun. 2015, 6, 1–9.
- Chiba, T.; Li, Y.H.; Yamane, T.; Ogikubo, O.; Fukuoka, M.; Arai, R.; Matsui, N. Inhibition of recombinant dipeptidyl peptidase III by synthetic hemorphin-like peptides. Peptides 2003, 24, 773–778.
- Jung, K.Y.; Moon, H.D.; Lee, G.E.; Lim, H.H.; Park, C.S.; Kim, Y.C. Structure- activity relationship studies of spinorphin as a potent and selective human P2X3 receptor antagonist. J. Med. Chem. 2007, 50, 4543–4547.
- Todorov, P.; Peneva, P.; Tchekalarova, J.; Rangelov, M.; Georgieva, S.; Todorova, N. Synthesis, characterization and anticonvulsant activity of new series of N-modified analogues of VV-Hemorphin-5 with aminophosphonate moiety. Amino Acids 2019, 51, 10–12.
- Dakubo, G.D. Brain Cancer Biomarkers in Proximal Fluids. In Cancer Biomarkers in Body Fluids; Springer: Cham, Switzerland, 2019; pp. 211–218.
- MacDonald, S.M.; Roscoe, S.G. Electrochemical oxidation reactions of tyrosine, tryptophan and related dipeptides. Electrochim. Acta 1997, 42, 1189–1200.
- Bernhardt, R. Cytochromes P450 as versatile biocatalysts. J. Biotechnol. 2006, 124, 128–145.


