 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Baptiste Maille | -- | 3957 | 2022-11-30 08:18:19 | | | |
| 2 | Lindsay Dong | Meta information modification | 3957 | 2022-12-01 02:00:05 | | |
Video Upload Options
Atrial fibrillation (AF) is a multifactorial sustained cardiac arrhythmia, and it is now considered a real worldwide public health issue. Despite the substantial progress that has been made in the detection and management of AF, the underlying molecular mechanisms associated with the onset of atrial fibrillation and its progression remain still unclear. Among these molecular mechanisms, the implication of the adenosinergic system in AF has increased, since the accumulation of experimental data suggests that the increase in the adenosine blood level and the remodeling expression of the adenosine receptors might be part of the AF pathophysiology. Unfortunately, the adenosinergic system still has a Janus face in cardiac arrythmias, since adenosine can have both antiarrhythmic or proarrhythmic actions, along with adenosine receptors, which can lead to either profibrotic or antifibrotic effects.
1. Adenosinergic System Signaling
1.1. Metabolism of Adenosine
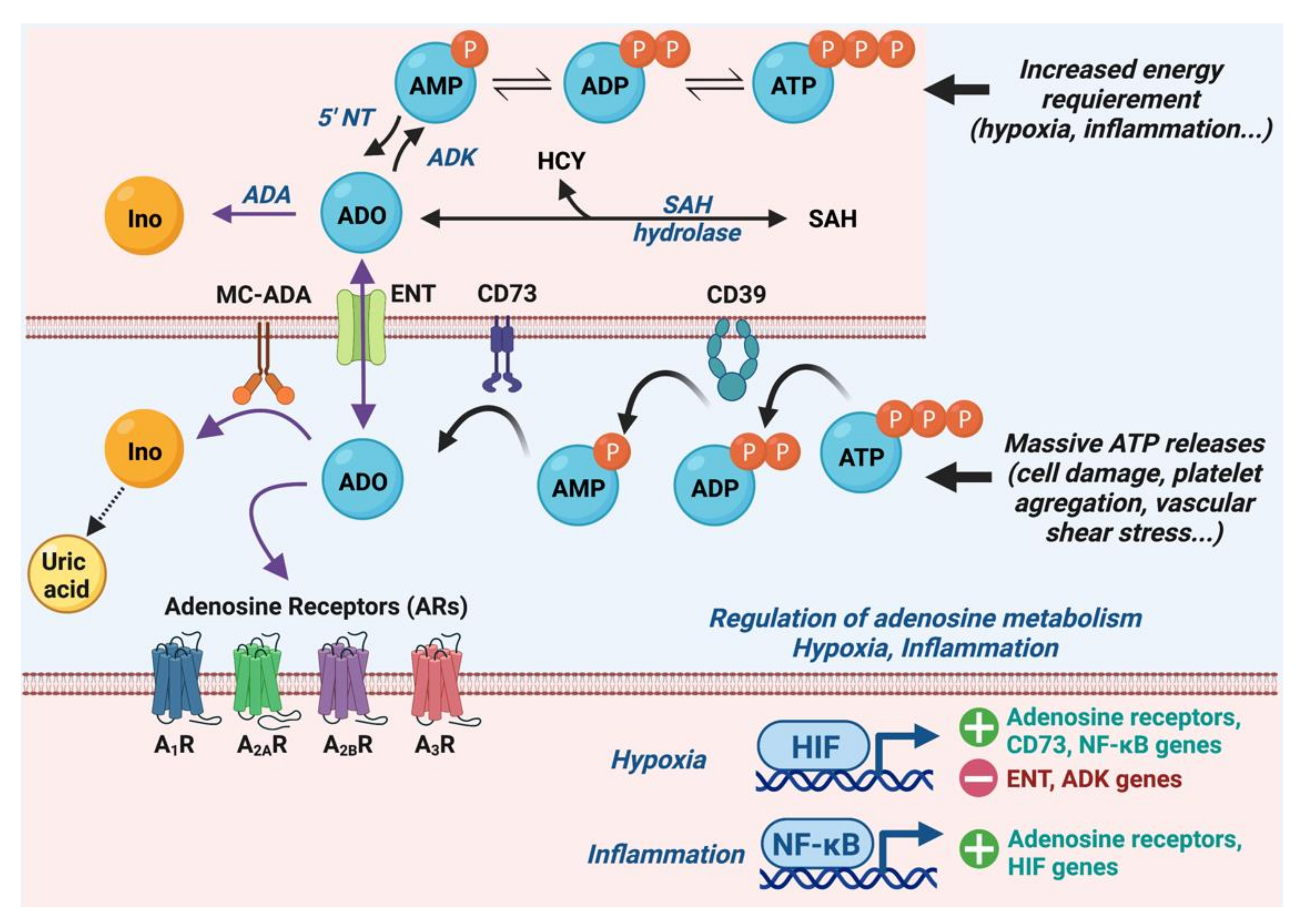
1.2. Adenosine Receptors and Their Cardiac Effects
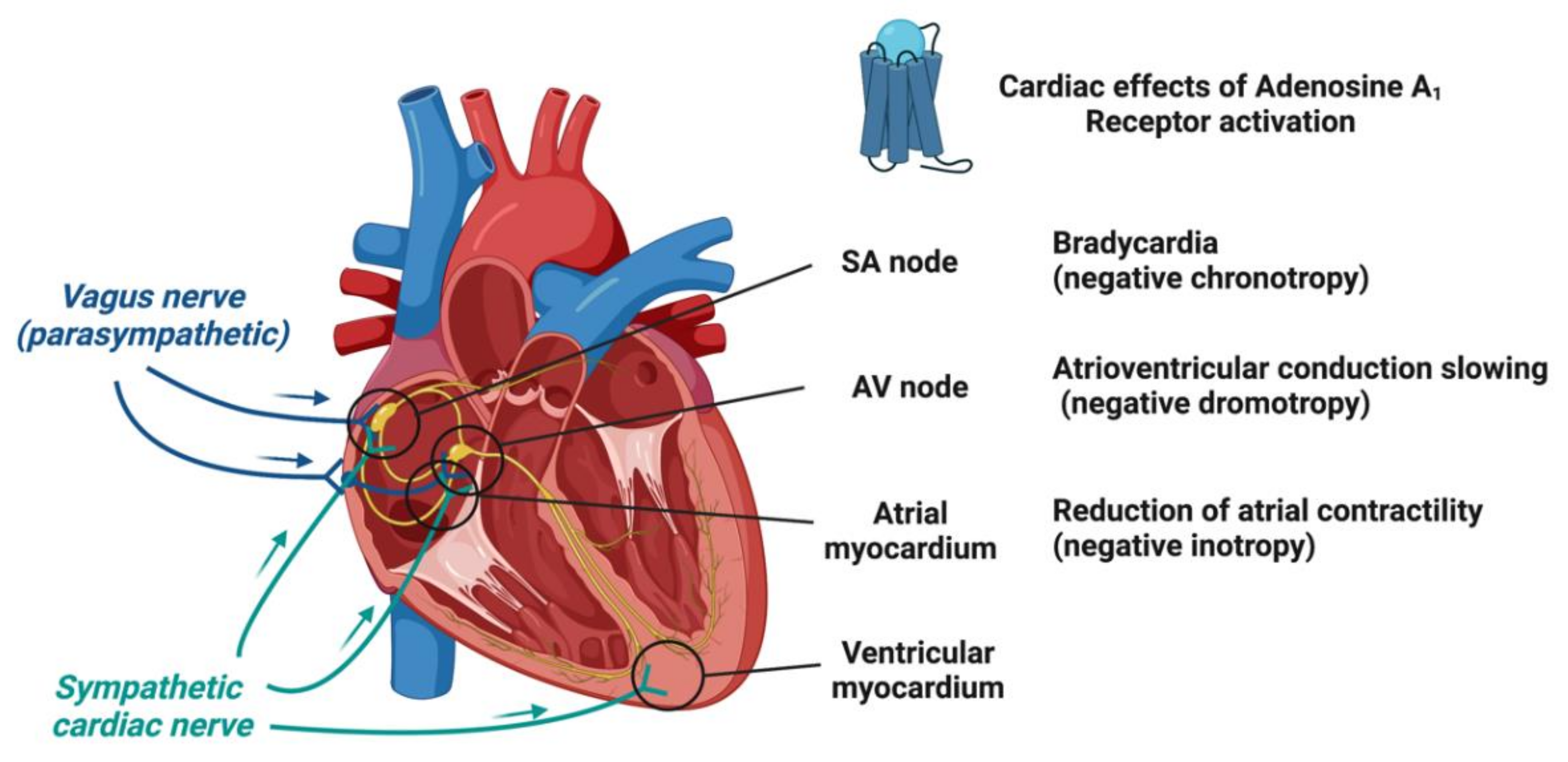
1.3. Molecular and Ionic Bases of Adenosine Effects in Cardiac Electrophysiology
1.3.1. Effects of Adenosine on Ionic Currents
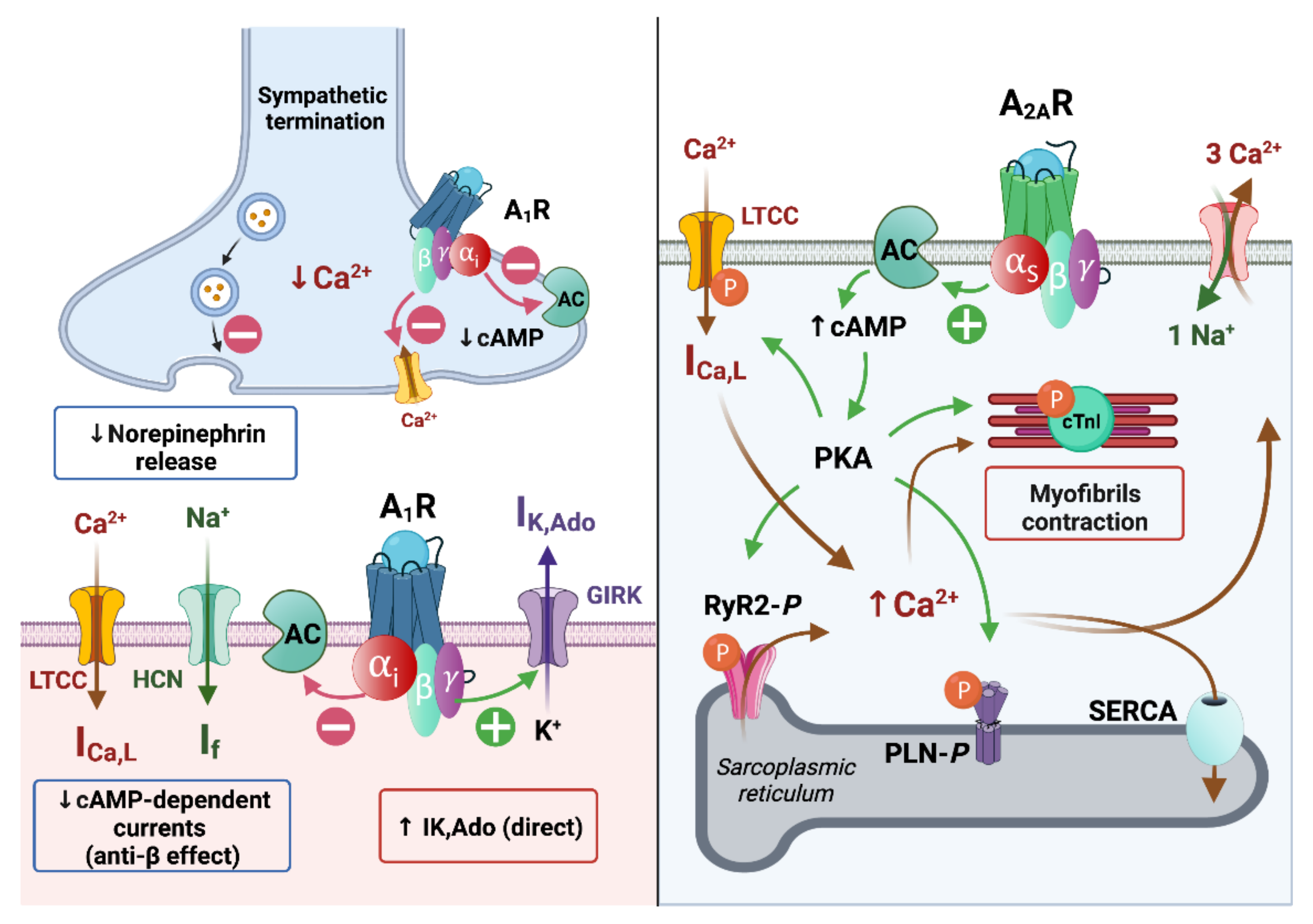
1.3.2. Adenosine Effects on the Action Potential in Nodal Cells and Working Cardiomyocytes
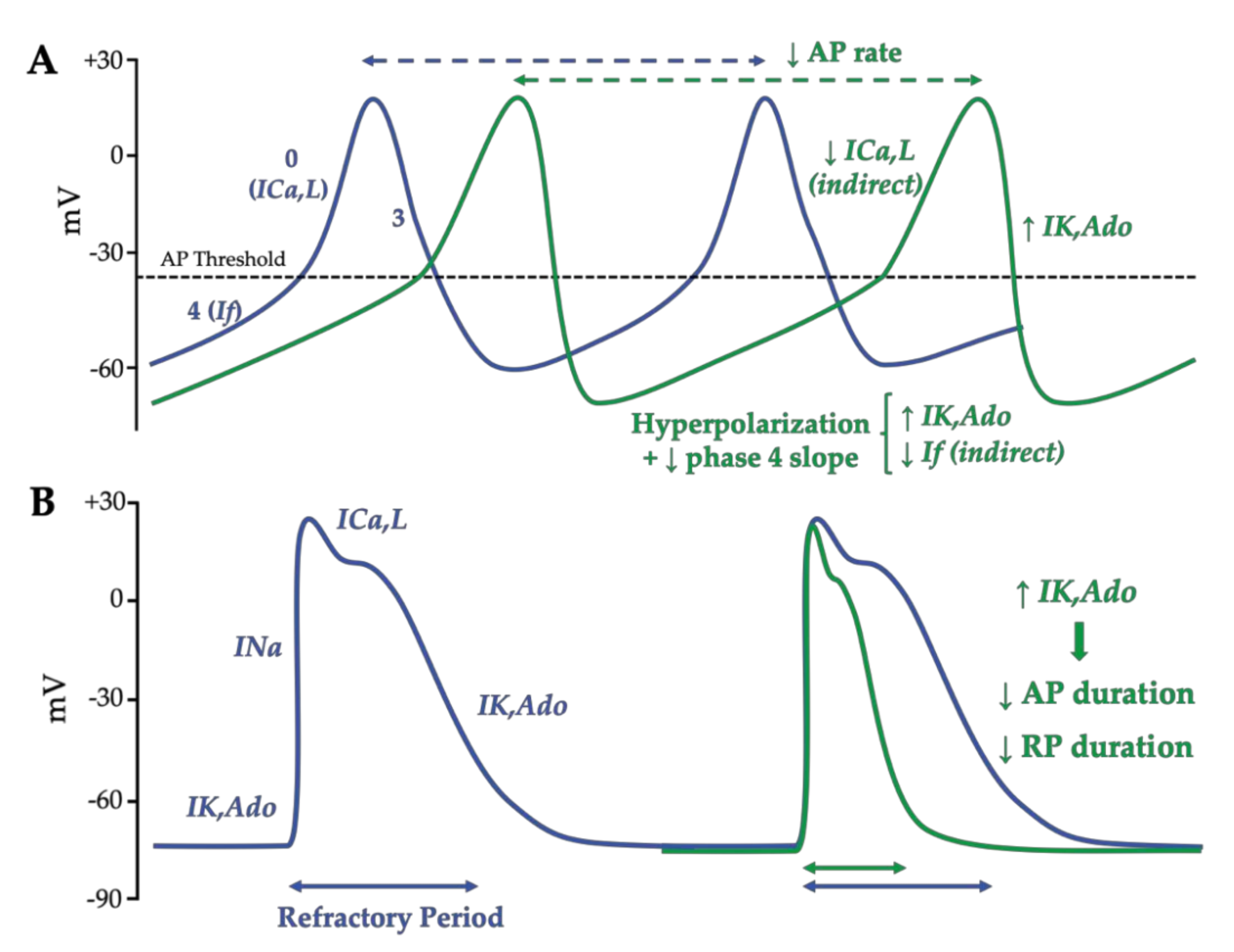
2. Pathophysiology of Atrial Fibrillation
2.1. Atrial Fibrillation Triggers
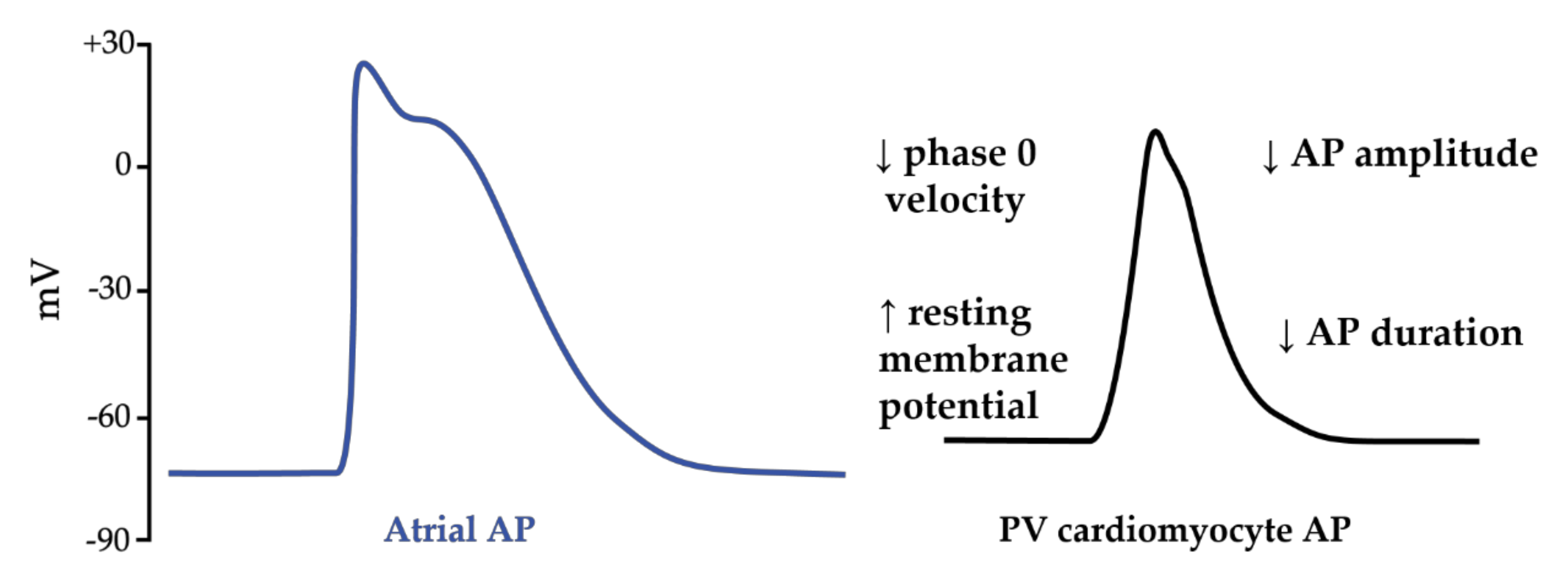
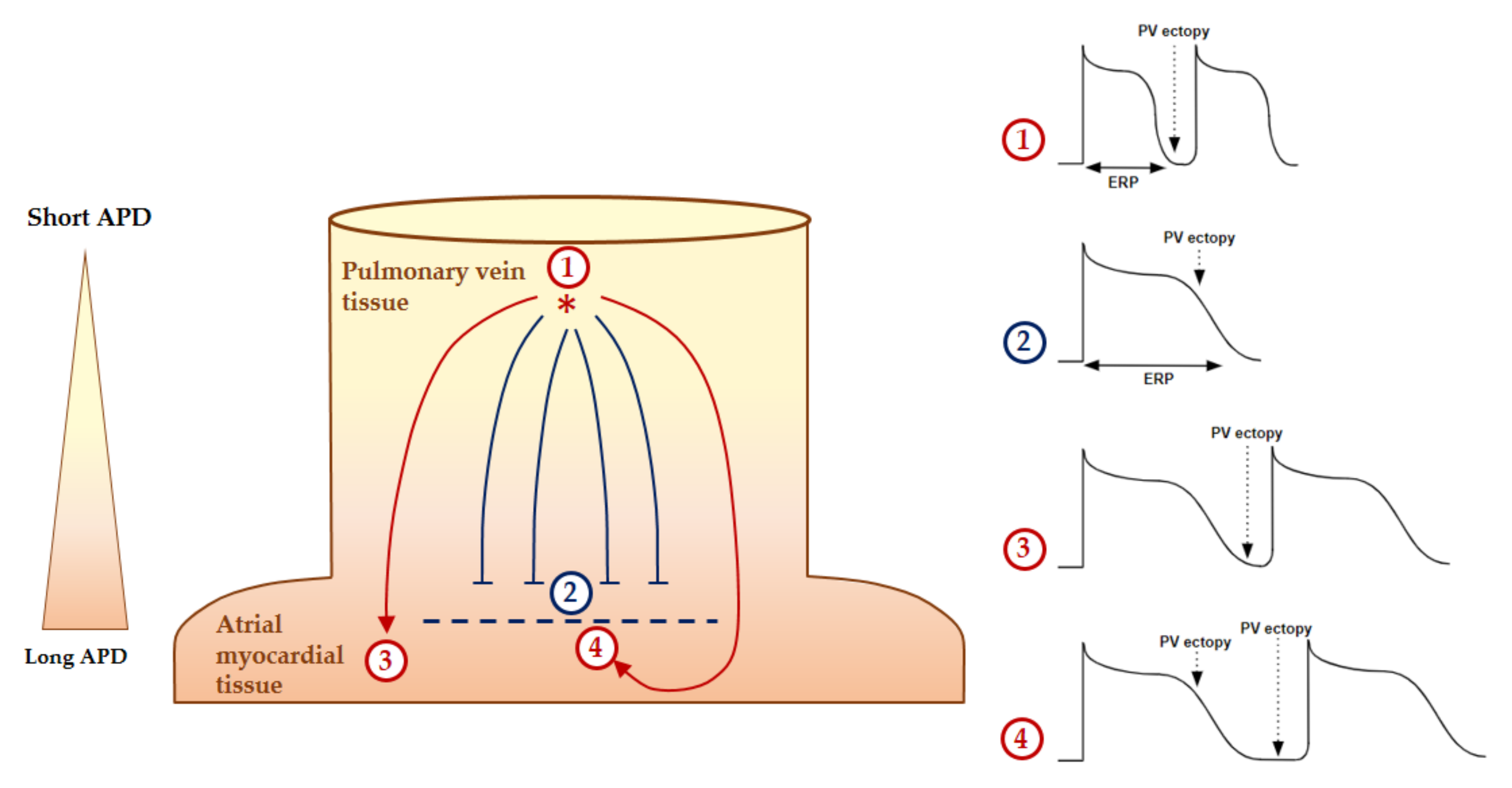
2.2. Mechanisms of AF Perpetuation
2.3. Substrate of Atrial Fibrillation and Atrial Cardiomyopathy
2.4. Pejorative Modulators of Atrial Fibrillation
3. Arrhythmogenic Effects of the Adenosinergic System
3.1. Adenosine Level and Expression of Adenosine Receptors in AF Patients
High adenosine plasma levels have been found in the left atria of patients during episodes of paroxysmal AF and in persistent AF [65]. The adenosine plasma concentrations then normalized after spontaneous or electrical cardioversion in sinus rhythm [65]. Moreover, the adenosine plasma concentrations in peripheric blood circulation were also higher in permanent AF compared to paroxysmal AF and controls [65]. The high adenosine plasma concentrations could be attributed to peripheral hypoxemia caused by the decrease in the left ventricular output in AF [65][66].
A high adenosine plasma concentration could also be a consequence of energy use in specific underlying cardiovascular conditions, including hypertension [67][68], chronic heart failure [69][70] or vagal syncope [71][72]. These are especially known to be AF risk factors. Interestingly, AF initiation has been described during the strong release of adenosine or the use of extrinsic adenosine injection [73][74].
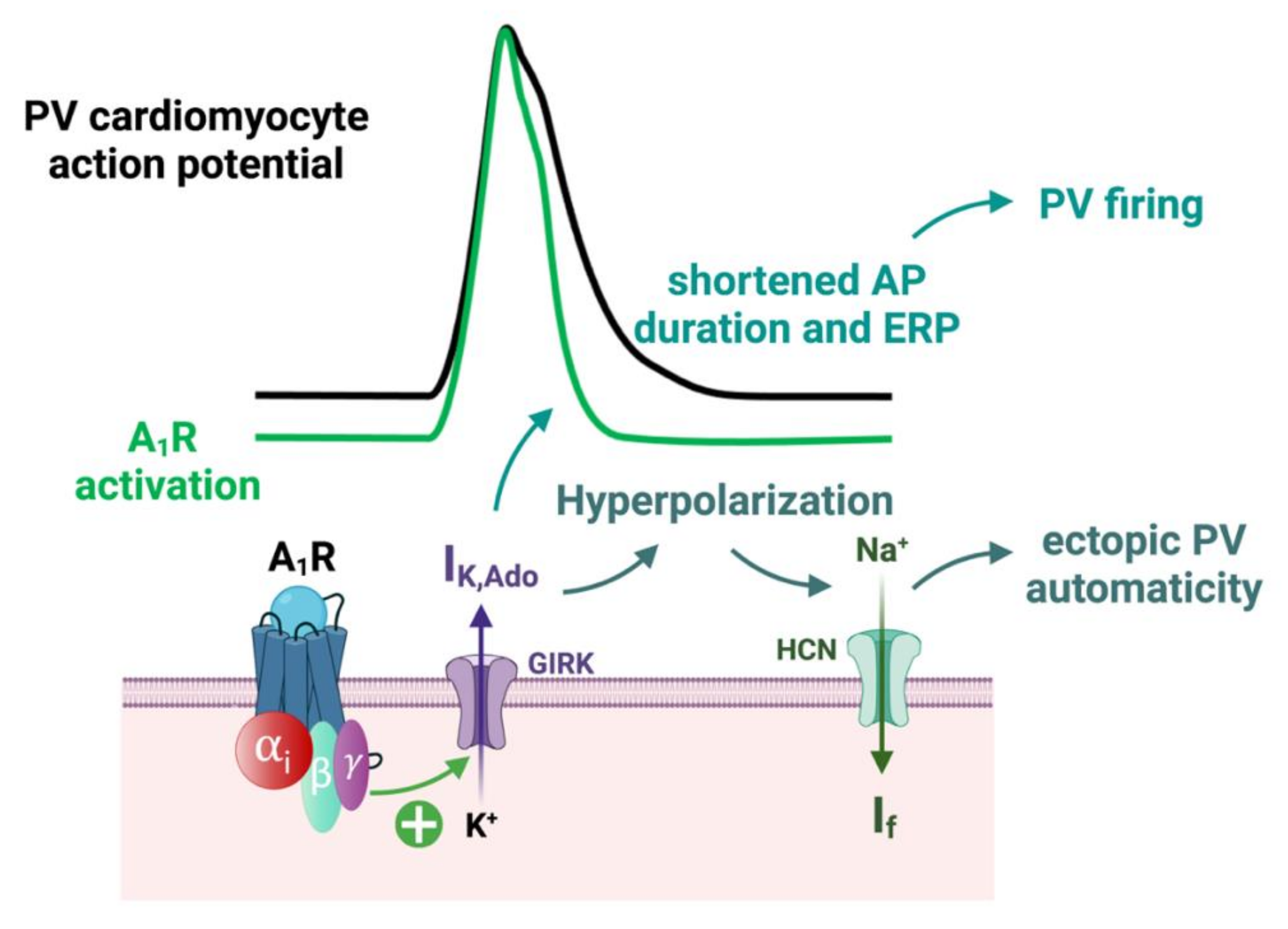
3.2. Implication of A1 Receptors in AF
3.3. Implication of AR in the Remodeling of Calcium Handling
3.4. Modulation of Atrial Fibrosis by A2B Receptors
4. Association between Atrial Fibrillation Risk Factors and the Adenosinergic System
References
- Drury, A.N.; Szent-Györgyi, A. The Physiological Activity of Adenine Compounds with Especial Reference to Their Action upon the Mammalian Heart. J. Physiol. 1929, 68, 213–237.
- Burnstock G Purinergic Nerves. Pharmacol. Rev. 1972, 24, 509–581.
- Szentmiklosi, A.J.; Galajda, Z.; Cseppento, Á.; Gesztelyi, R.; Susán, Z.; Hegyi, B.; Nánási, P.P. The Janus Face of Adenosine: Antiarrhythmic and Proarrhythmic Actions. Curr. Pharm. Des. 2015, 21, 965–976.
- Guieu, R.; Deharo, J.-C.; Maille, B.; Crotti, L.; Torresani, E.; Brignole, M.; Parati, G. Adenosine and the Cardiovascular System: The Good and the Bad. J. Clin. Med. 2020, 9, E1366.
- Fredholm, B.B. Adenosine--a Physiological or Pathophysiological Agent? J. Mol. Med. 2014, 92, 201–206.
- Deussen, A.; Lloyd, H.G.; Schrader, J. Contribution of S-Adenosylhomocysteine to Cardiac Adenosine Formation. J. Mol. Cell. Cardiol. 1989, 21, 773–782.
- Sumi, Y.; Woehrle, T.; Chen, Y.; Yao, Y.; Li, A.; Junger, W.G. Adrenergic Receptor Activation Involves ATP Release and Feedback through Purinergic Receptors. Am. J. Physiol. Cell Physiol. 2010, 299, C1118–C1126.
- Le, G.Y.; Essackjee, H.C.; Ballard, H.J. Intracellular Adenosine Formation and Release by Freshly-Isolated Vascular Endothelial Cells from Rat Skeletal Muscle: Effects of Hypoxia and/or Acidosis. Biochem. Biophys. Res. Commun. 2014, 450, 93–98.
- Colgan, S.P.; Eltzschig, H.K.; Eckle, T.; Thompson, L.F. Physiological Roles for Ecto-5’-Nucleotidase (CD73). Purinerg. Signal. 2006, 2, 351–360.
- Fredholm, B.B.; Arslan, G.; Halldner, L.; Kull, B.; Schulte, G.; Wasserman, W. Structure and Function of Adenosine Receptors and Their Genes. Naunyn. Schmiedebergs. Arch. Pharmacol. 2000, 362, 364–374.
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and Classification of Adenosine Receptors. Pharmacol. Rev. 2001, 53, 527–552.
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625.
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228.
- Headrick, J.P.; Ashton, K.J.; Rose’meyer, R.B.; Peart, J.N. Cardiovascular Adenosine Receptors: Expression, Actions and Interactions. Pharmacol. Ther. 2013, 140, 92–111.
- Chandrasekera, P.C.; McIntosh, V.J.; Cao, F.X.; Lasley, R.D. Differential Effects of Adenosine A2a and A2b Receptors on Cardiac Contractility. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H2082–H2089.
- Musser, B.; Morgan, M.E.; Leid, M.; Murray, T.F.; Linden, J.; Vestal, R.E. Species Comparison of Adenosine and Beta-Adrenoceptors in Mammalian Atrial and Ventricular Myocardium. Eur. J. Pharmacol. 1993, 246, 105–111.
- Shryock, J.C.; Belardinelli, L. Adenosine and Adenosine Receptors in the Cardiovascular System: Biochemistry, Physiology, and Pharmacology. Am. J. Cardiol. 1997, 79, 2–10.
- Schrader, J.; Baumann, G.; Gerlach, E. Adenosine as Inhibitor of Myocardial Effects of Catecholamines. Pflugers. Arch. 1977, 372, 29–35.
- Dobson, J.G. Mechanism of Adenosine Inhibition of Catecholamine-Induced Responses in Heart. Circ. Res. 1983, 52, 151–160.
- Wennmalm, M.; Fredholm, B.B.; Hedqvist, P. Adenosine as a Modulator of Sympathetic Nerve-Stimulation-Induced Release of Noradrenaline from the Isolated Rabbit Heart. Acta. Physiol. Scand. 1988, 132, 487–494.
- Belardinelli, L.; Giles, W.R.; West, A. Ionic Mechanisms of Adenosine Actions in Pacemaker Cells from Rabbit Heart. J. Physiol. 1988, 405, 615–633.
- Lerman, B.B.; Markowitz, S.M.; Cheung, J.W.; Liu, C.F.; Thomas, G.; Ip, J.E. Supraventricular Tachycardia. Circ. Arrhythmia Elec. 2018, 11, e006953.
- Brugada, J.; Katritsis, D.G.; Arbelo, E.; Arribas, F.; Bax, J.J.; Blomström-Lundqvist, C.; Calkins, H.; Corrado, D.; Deftereos, S.G.; Diller, G.-P.; et al. 2019 ESC Guidelines for the Management of Patients with Supraventricular TachycardiaThe Task Force for the Management of Patients with Supraventricular Tachycardia of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 41, 655–720.
- Lokhandwala, M.F. Inhibition of Cardiac Sympathetic Neurotransmission by Adenosine. Eur. J. Pharmacol. 1979, 60, 353–357.
- Belardinelli, L.; Shryock, J.C.; Song, Y.; Wang, D.; Srinivas, M. Ionic Basis of the Electrophysiological Actions of Adenosine on Cardiomyocytes. FASEB J. 1995, 9, 359–365.
- DiFrancesco, D.; Tortora, P. Direct Activation of Cardiac Pacemaker Channels by Intracellular Cyclic AMP. Nature 1991, 351, 145–147.
- Wang, X.; Liang, B.; Skibsbye, L.; Olesen, S.-P.; Grunnet, M.; Jespersen, T. GIRK Channel Activation via Adenosine or Muscarinic Receptors Has Similar Effects on Rat Atrial Electrophysiology. J. Cardiovasc. Pharmacol. 2013, 62, 192–198.
- Pfaffinger, P.J.; Martin, J.M.; Hunter, D.D.; Nathanson, N.M.; Hille, B. GTP-Binding Proteins Couple Cardiac Muscarinic Receptors to a K Channel. Nature 1985, 317, 536–538.
- Lopatin, A.N.; Nichols, C.G. Inward Rectifiers in the Heart: An Update on I(K1). J. Mol. Cell. Cardiol. 2001, 33, 625–638.
- Anumonwo, J.M.B.; Lopatin, A.N. Cardiac Strong Inward Rectifier Potassium Channels. J. Mol. Cell. Cardiol. 2010, 48, 45–54.
- Guo, D.; Ramu, Y.; Klem, A.M.; Lu, Z. Mechanism of Rectification in Inward-Rectifier K+ Channels. J. Gen. Physiol. 2003, 121, 261–276.
- Kilpatrick, E.L.; Narayan, P.; Mentzer, R.M.; Lasley, R.D. Cardiac Myocyte Adenosine A2a Receptor Activation Fails to Alter CAMP or Contractility: Role of Receptor Localization. Am. J. Physiol. Heart. Circ. Physiol. 2002, 282, H1035–H1040.
- Boknik, P.; Drzewiecki, K.; Eskandar, J.; Gergs, U.; Grote-Wessels, S.; Fabritz, L.; Kirchhof, P.; Müller, F.U.; Stümpel, F.; Schmitz, W.; et al. Phenotyping of Mice with Heart Specific Overexpression of A2A-Adenosine Receptors: Evidence for Cardioprotective Effects of A2A-Adenosine Receptors. Front. Pharmacol. 2018, 9, 13.
- Hove-Madsen, L.; Prat-Vidal, C.; Llach, A.; Ciruela, F.; Casadó, V.; Lluis, C.; Bayes-Genis, A.; Cinca, J.; Franco, R. Adenosine A2A Receptors Are Expressed in Human Atrial Myocytes and Modulate Spontaneous Sarcoplasmic Reticulum Calcium Release. Cardiovasc. Res. 2006, 72, 292–302.
- Llach, A.; Molina, C.E.; Prat-Vidal, C.; Fernandes, J.; Casadó, V.; Ciruela, F.; Lluís, C.; Franco, R.; Cinca, J.; Hove-Madsen, L. Abnormal calcium handling in atrial fibrillation is linked to up-regulation of adenosine A2A receptors. Eur. Heart J. 2011, 32, 721–729.
- Boknik, P.; Eskandar, J.; Hofmann, B.; Zimmermann, N.; Neumann, J.; Gergs, U. Role of Cardiac A2A Receptors Under Normal and Pathophysiological Conditions. Front. Pharmacol. 2020, 11, 627838.
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195.
- Brandenburg, S.; Pawlowitz, J.; Steckmeister, V.; Subramanian, H.; Uhlenkamp, D.; Scardigli, M.; Mushtaq, M.; Amlaz, S.I.; Kohl, T.; Wegener, J.W.; et al. A Junctional CAMP Compartment Regulates Rapid Ca2+ Signaling in Atrial Myocytes. J. Mol. Cell. Cardiol. 2022, 165, 141–157.
- Christ, T.; Boknik, P.; Wöhrl, S.; Wettwer, E.; Graf, E.M.; Bosch, R.F.; Knaut, M.; Schmitz, W.; Ravens, U.; Dobrev, D. L-Type Ca2+ Current Downregulation in Chronic Human Atrial Fibrillation Is Associated With Increased Activity of Protein Phosphatases. Circulation 2004, 110, 2651–2657.
- Bokník, P.; Unkel, C.; Kirchhefer, U.; Kleideiter, U.; Klein-Wiele, O.; Knapp, J.; Linck, B.; Lüss, H.; Ulrich Müller, F.; Schmitz, W.; et al. Regional Expression of Phospholamban in the Human Heart. Cardiovasc. Res. 1999, 43, 67–76.
- Dhamoon, A.S.; Pandit, S.V.; Sarmast, F.; Parisian, K.R.; Guha, P.; Li, Y.; Bagwe, S.; Taffet, S.M.; Anumonwo, J.M.B. Unique Kir2.x Properties Determine Regional and Species Differences in the Cardiac Inward Rectifier K+ Current. Circ. Res. 2004, 94, 1332–1339.
- Chandler, N.J.; Greener, I.D.; Tellez, J.O.; Inada, S.; Musa, H.; Molenaar, P.; Difrancesco, D.; Baruscotti, M.; Longhi, R.; Anderson, R.H.; et al. Molecular Architecture of the Human Sinus Node: Insights into the Function of the Cardiac Pacemaker. Circulation 2009, 119, 1562–1575.
- MacDonald, E.A.; Rose, R.A.; Quinn, T.A. Neurohumoral Control of Sinoatrial Node Activity and Heart Rate: Insight From Experimental Models and Findings From Humans. Front. Physiol. 2020, 11, 170.
- Kléber, A.G.; Rudy, Y. Basic Mechanisms of Cardiac Impulse Propagation and Associated Arrhythmias. Physiol. Rev. 2004, 84, 431–488.
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the Diagnosis and Management of Atrial Fibrillation Developed in Collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2021, 42, 373–498.
- Cheniti, G.; Vlachos, K.; Pambrun, T.; Hooks, D.; Frontera, A.; Takigawa, M.; Bourier, F.; Kitamura, T.; Lam, A.; Martin, C.; et al. Atrial Fibrillation Mechanisms and Implications for Catheter Ablation. Front. Physiol. 2018, 9, 1458.
- Haïssaguerre, M.; Jaïs, P.; Shah, D.C.; Takahashi, A.; Hocini, M.; Quiniou, G.; Garrigue, S.; Le Mouroux, A.; Le Métayer, P.; Clémenty, J. Spontaneous Initiation of Atrial Fibrillation by Ectopic Beats Originating in the Pulmonary Veins. N. Engl. J. Med. 1998, 339, 659–666.
- Lin, W.-S.; Tai, C.-T.; Hsieh, M.-H.; Tsai, C.-F.; Lin, Y.-K.; Tsao, H.-M.; Huang, J.-L.; Yu, W.-C.; Yang, S.-P.; Ding, Y.-A.; et al. Catheter Ablation of Paroxysmal Atrial Fibrillation Initiated by Non–Pulmonary Vein Ectopy. Circulation 2003, 107, 3176–3183.
- Lee, S.-H.; Tai, C.-T.; Hsieh, M.-H.; Tsao, H.-M.; Lin, Y.-J.; Chang, S.-L.; Huang, J.-L.; Lee, K.-T.; Chen, Y.-J.; Cheng, J.-J.; et al. Predictors of Non-Pulmonary Vein Ectopic Beats Initiating Paroxysmal Atrial Fibrillation: Implication for Catheter Ablation. J. Am. Coll. Cardiol. 2005, 46, 1054–1059.
- Santangeli, P.; Marchlinski, F.E. Techniques for the Provocation, Localization, and Ablation of Non–Pulmonary Vein Triggers for Atrial Fibrillation. Heart Rhythm 2017, 14, 1087–1096.
- Arentz, T.; Haegeli, L.; Sanders, P.; Weber, R.; Neumann, F.J.; Kalusche, D.; Haïssaguerre, M. High-Density Mapping of Spontaneous Pulmonary Vein Activity Initiating Atrial Fibrillation in Humans. J. Cardiovasc. Electrophysiol. 2007, 18, 31–38.
- Ehrlich, J.R.; Cha, T.-J.; Zhang, L.; Chartier, D.; Melnyk, P.; Hohnloser, S.H.; Nattel, S. Cellular Electrophysiology of Canine Pulmonary Vein Cardiomyocytes: Action Potential and Ionic Current Properties. J. Physiol. 2003, 551, 801–813.
- Haissaguerre, M.; Shah, A.J.; Cochet, H.; Hocini, M.; Dubois, R.; Efimov, I.; Vigmond, E.; Bernus, O.; Trayanova, N. Intermittent Drivers Anchoring to Structural Heterogeneities as a Major Pathophysiological Mechanism of Human Persistent Atrial Fibrillation. J. Physiol. 2016, 594, 2387–2398.
- Qu, Z. Critical Mass Hypothesis Revisited: Role of Dynamical Wave Stability in Spontaneous Termination of Cardiac Fibrillation. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H255–H263.
- Hansen, B.J.; Csepe, T.A.; Zhao, J.; Ignozzi, A.J.; Hummel, J.D.; Fedorov, V.V. Maintenance of Atrial Fibrillation: Are Reentrant Drivers With Spatial Stability the Key? Circ. Arrhythm. Electrophysiol. 2016, 9, e004398.
- Lim, H.S.; Hocini, M.; Dubois, R.; Denis, A.; Derval, N.; Zellerhoff, S.; Yamashita, S.; Berte, B.; Mahida, S.; Komatsu, Y.; et al. Complexity and Distribution of Drivers in Relation to Duration of Persistent Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 69, 1257–1269.
- Roney, C.H.; Bayer, J.D.; Cochet, H.; Meo, M.; Dubois, R.; Jaïs, P.; Vigmond, E.J. Variability in Pulmonary Vein Electrophysiology and Fibrosis Determines Arrhythmia Susceptibility and Dynamics. PLOS Comput. Biol. 2018, 14, e1006166.
- Frontera, A.; Pagani, S.; Limite, L.R.; Peirone, A.; Fioravanti, F.; Enache, B.; Cuellar, S.J.; Vlachos, K.; Meyer, C.; Montesano, G.; et al. Slow Conduction Corridors and Pivot Sites Characterize the Electrical Remodeling in Atrial Fibrillation. JACC Cli. Electrophysiol. 2022, 8, 561–577.
- Allessie, M.A.; Bonke, F.I.; Schopman, F.J. Circus Movement in Rabbit Atrial Muscle as a Mechanism of Tachycardia. III. The “Leading Circle” Concept: A New Model of Circus Movement in Cardiac Tissue without the Involvement of an Anatomical Obstacle. Circ. Res. 1977, 41, 9–18.
- Nattel, S.; Xiong, F.; Aguilar, M. Demystifying Rotors and Their Place in Clinical Translation of Atrial Fibrillation Mechanisms. Nat. Rev. Cardiol. 2017, 14, 509–520.
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological Mechanisms of Atrial Fibrillation: A Translational Appraisal. Physiol. Rev. 2011, 91, 265–325.
- Maille, B.; Das, M.; Hussein, A.; Shaw, M.; Chaturvedi, V.; Williams, E.; Morgan, M.; Ronayne, C.; Snowdon, R.L.; Gupta, D. Reverse Electrical and Structural Remodeling of the Left Atrium Occurs Early after Pulmonary Vein Isolation for Persistent Atrial Fibrillation. J. Interv. Card. Electrophysiol. 2020, 58, 9–19.
- Goette, A.; Kalman, J.M.; Aguinaga, L.; Akar, J.; Cabrera, J.A.; Chen, S.A.; Chugh, S.S.; Corradi, D.; D’Avila, A.; Dobrev, D.; et al. EHRA/HRS/APHRS/SOLAECE Expert Consensus on Atrial Cardiomyopathies: Definition, Characterization, and Clinical Implication. EP Europace 2016, 18, 1455–1490.
- Heijman, J.; Guichard, J.-B.; Dobrev, D.; Nattel, S. Translational Challenges in Atrial Fibrillation. Circ. Res. 2018, 122, 752–773.
- Maille, B.; Marlinge, M.; Vairo, D.; Mottola, G.; Koutbi, L.; Deharo, P.; Gastaldi, M.; Gaudry, M.; Guiol, C.; Bottone, S.; et al. Adenosine Plasma Level in Patients with Paroxysmal or Persistent Atrial Fibrillation and Normal Heart during Ablation Procedure and/or Cardioversion. Purinergic Signal. 2019, 15, 45–52.
- Cha, Y.-M.; Dzeja, P.P.; Shen, W.K.; Jahangir, A.; Hart, C.Y.T.; Terzic, A.; Redfield, M.M. Failing Atrial Myocardium: Energetic Deficits Accompany Structural Remodeling and Electrical Instability. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1313–H1320.
- Zhang, W.; Zhang, Y.; Wang, W.; Dai, Y.; Ning, C.; Luo, R.; Sun, K.; Glover, L.; Grenz, A.; Sun, H.; et al. Elevated Ecto-5’-Nucleotidase-Mediated Increased Renal Adenosine Signaling via A2B Adenosine Receptor Contributes to Chronic Hypertension. Circ. Res. 2013, 112, 1466–1478.
- Silhol, F.; Marlinge, M.; Guiol, C.; Chefrour, M.; Mace, P.; Criado, C.; Kipson, N.; Vaisse, B.; Vairo, D.; Sarlon, G.; et al. Characterization of Adenosine A2 Receptors in Peripheral Blood Mononuclear Cells of Patients with Fibromuscular Dysplasia. Hypertens. Res. 2020, 43, 466–469.
- Gaubert, M.; Marlinge, M.; Kerbaul, F.; Resseguier, N.; Laine, M.; Cautella, J.; Cordier, C.; Colomb, B.; Kipson, N.; Thuny, F.; et al. Adenosine Plasma Level and A2A Receptor Expression in Patients With Cardiogenic Shock. Crit. Care Med. 2018, 46, e874–e880.
- Franceschi, F.; Deharo, J.-C.; Giorgi, R.; By, Y.; Monserrat, C.; Condo, J.; Ibrahim, Z.; Saadjian, A.; Guieu, R. Peripheral Plasma Adenosine Release in Patients with Chronic Heart Failure. Heart 2009, 95, 651–655.
- Linz, D.; Elliott, A.D.; Hohl, M.; Malik, V.; Schotten, U.; Dobrev, D.; Nattel, S.; Böhm, M.; Floras, J.; Lau, D.H.; et al. Role of Autonomic Nervous System in Atrial Fibrillation. Int. J. Cardiol. 2019, 287, 181–188.
- Saadjian, A.Y.; Lévy, S.; Franceschi, F.; Zouher, I.; Paganelli, F.; Guieu, R.P. Role of Endogenous Adenosine as a Modulator of Syncope Induced during Tilt Testing. Circulation 2002, 106, 569–574.
- Tebbenjohanns, J.; Schumacher, B.; Pfeiffer, D.; Jung, W.; Lüderitz, B. Dose and Rate-Dependent Effects of Adenosine on Atrial Action Potential Duration in Humans. J. Interv. Card. Electrophysiol. 1997, 1, 33–37.
- Strickberger, S.A.; Man, K.C.; Daoud, E.G.; Goyal, R.; Brinkman, K.; Knight, B.P.; Weiss, R.; Bahu, M.; Morady, F. Adenosine-Induced Atrial Arrhythmia: A Prospective Analysis. Ann. Intern. Med. 1997, 127, 417–422.
- Soattin, L.; Lubberding, A.F.; Bentzen, B.H.; Christ, T.; Jespersen, T. Inhibition of Adenosine Pathway Alters Atrial Electrophysiology and Prevents Atrial Fibrillation. Front. Physiol. 2020, 11, 493.
- Van Wagoner, D.R.; Pond, A.L.; Lamorgese, M.; Rossie, S.S.; McCarthy, P.M.; Nerbonne, J.M. Atrial L-Type Ca2+ Currents and Human Atrial Fibrillation. Circ. Res. 1999, 85, 428–436.
- Reinhardt, F.; Beneke, K.; Pavlidou, N.G.; Conradi, L.; Reichenspurner, H.; Hove-Madsen, L.; Molina, C.E. Abnormal Calcium Handling in Atrial Fibrillation Is Linked to Changes in Cyclic AMP Dependent Signaling. Cells 2021, 10, 3042.
- Lehnart, S.E.; Wehrens, X.H.T.; Laitinen, P.J.; Reiken, S.R.; Deng, S.-X.; Cheng, Z.; Landry, D.W.; Kontula, K.; Swan, H.; Marks, A.R. Sudden Death in Familial Polymorphic Ventricular Tachycardia Associated with Calcium Release Channel (Ryanodine Receptor). Leak. Circulation 2004, 109, 3208–3214.
- Burashnikov, A.; Antzelevitch, C. Reinduction of Atrial Fibrillation Immediately after Termination of the Arrhythmia Is Mediated by Late Phase 3 Early Afterdepolarization-Induced Triggered Activity. Circulation 2003, 107, 2355–2360.
- Visentin, S.; Wu, S.N.; Belardinelli, L. Adenosine-Induced Changes in Atrial Action Potential: Contribution of Ca and K Currents. Am. J. Physiol. 1990, 258, H1070–H1078.
- Vecchio, E.A.; White, P.J.; May, L.T. Targeting Adenosine Receptors for the Treatment of Cardiac Fibrosis. Front. Pharmacol. 2017, 8, 243.
- Vecchio, E.A.; White, P.J.; May, L.T. The Adenosine A2B G Protein-Coupled Receptor: Recent Advances and Therapeutic Implications. Pharmacol. Ther. 2019, 198, 20–33.
- Toldo, S.; Zhong, H.; Mezzaroma, E.; Van Tassell, B.W.; Kannan, H.; Zeng, D.; Belardinelli, L.; Voelkel, N.F.; Abbate, A. GS-6201, a Selective Blocker of the A2B Adenosine Receptor, Attenuates Cardiac Remodeling after Acute Myocardial Infarction in the Mouse. J. Pharmacol. Exp. Ther. 2012, 343, 587–595.
- Zhong, H.; Belardinelli, L.; Zeng, D. Pro-Fibrotic Role of the A2B Adenosine Receptor in Human Cardiac Fibroblasts. J. Card. Fail. 2011, 17, S65.
- Lu, D.; Insel, P.A. Hydrolysis of Extracellular ATP by Ectonucleoside Triphosphate Diphosphohydrolase (ENTPD) Establishes the Set Point for Fibrotic Activity of Cardiac Fibroblasts. J. Biol. Chem. 2013, 288, 19040–19049.
- Phosri, S.; Bunrukchai, K.; Parichatikanond, W.; Sato, V.H.; Mangmool, S. Epac Is Required for Exogenous and Endogenous Stimulation of Adenosine A2B Receptor for Inhibition of Angiotensin II-Induced Collagen Synthesis and Myofibroblast Differentiation. Purinergic Signal. 2018, 14, 141–156.
- Chen, Y.; Epperson, S.; Makhsudova, L.; Ito, B.; Suarez, J.; Dillmann, W.; Villarreal, F. Functional Effects of Enhancing or Silencing Adenosine A2b Receptors in Cardiac Fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2478–H2486.
- Wakeno, M.; Minamino, T.; Seguchi, O.; Okazaki, H.; Tsukamoto, O.; Okada, K.; Hirata, A.; Fujita, M.; Asanuma, H.; Kim, J.; et al. Long-Term Stimulation of Adenosine A2b Receptors Begun After Myocardial Infarction Prevents Cardiac Remodeling in Rats. Circulation 2006, 114, 1923–1932.
- Maas, J.E.; Wan, T.C.; Figler, R.A.; Gross, G.J.; Auchampach, J.A. Evidence That the Acute Phase of Ischemic Preconditioning Does Not Require Signaling by the A2B Adenosine Receptor. J. Mol. Cell. Cardiol. 2010, 49, 886–893.
- Feng, W.; Song, Y.; Chen, C.; Lu, Z.Z.; Zhang, Y. Stimulation of Adenosine A2B Receptors Induces Interleukin-6 Secretion in Cardiac Fibroblasts via the PKC-Delta-P38 Signalling Pathway. Br. J. Pharmacol. 2010, 159, 1598–1607.
- Wragg, E.S.; Pannucci, P.; Hill, S.J.; Woolard, J.; Cooper, S.L. Involvement of β-Adrenoceptors in the Cardiovascular Responses Induced by Selective Adenosine A2A and A2B Receptor Agonists. Pharmacol. Res. Perspect. 2022, 10, e00975.
- Lee, S.W.; Anderson, A.; Guzman, P.A.; Nakano, A.; Tolkacheva, E.G.; Wickman, K. Atrial GIRK Channels Mediate the Effects of Vagus Nerve Stimulation on Heart Rate Dynamics and Arrhythmogenesis. Front. Physiol. 2018, 9, 943.
- Javed, S.; Gupta, D.; Lip, G.Y.H. Obesity and Atrial Fibrillation: Making Inroads through Fat. Eur. Heart J. Cardiovasc. Pharmacother. 2021, 7, 59–67.
- Nalliah, C.J.; Wong, G.R.; Lee, G.; Voskoboinik, A.; Kee, K.; Goldin, J.; Watts, T.; Linz, D.; Wirth, D.; Parameswaran, R.; et al. Sleep Apnoea Has a Dose-Dependent Effect on Atrial Remodelling in Paroxysmal but Not Persistent Atrial Fibrillation: A High-Density Mapping Study. EP Europace 2021, 23, 691–700.
- Sidhu, K.; Tang, A. Modifiable Risk Factors in Atrial Fibrillation: The Role of Alcohol, Obesity, and Sleep Apnea. Can. J. Cardiol. 2017, 33, 947–949.
- Sheng, Y.; Li, M.; Xu, M.; Zhang, Y.; Xu, J.; Huang, Y.; Li, X.; Yao, G.; Sui, W.; Zhang, M.; et al. Left Ventricular and Atrial Remodelling in Hypertensive Patients Using Thresholds from International Guidelines and EMINCA Data. Eur. Heart J. Cardiovasc. Imag. 2022, 23, 166–174.
- Sharifov, O.F.; Fedorov, V.V.; Beloshapko, G.G.; Glukhov, A.V.; Yushmanova, A.V.; Rosenshtraukh, L.V. Roles of Adrenergic and Cholinergic Stimulation in Spontaneous Atrial Fibrillation in Dogs. J. Am. Coll. Cardiol. 2004, 43, 483–490.
- Huang, J.L.; Wen, Z.C.; Lee, W.L.; Chang, M.S.; Chen, S.A. Changes of Autonomic Tone before the Onset of Paroxysmal Atrial Fibrillation. Int. J. Cardiol. 1998, 66, 275–283.
- Tan, A.Y.; Zhou, S.; Ogawa, M.; Song, J.; Chu, M.; Li, H.; Fishbein, M.C.; Lin, S.-F.; Chen, L.S.; Chen, P.-S. Neural Mechanisms of Paroxysmal Atrial Fibrillation and Paroxysmal Atrial Tachycardia in Ambulatory Canines. Circulation 2008, 118, 916–925.

