Atrial fibrillation (AF) is a multifactorial sustained cardiac arrhythmia, and it is now considered a real worldwide public health issue. Despite the substantial progress that has been made in the detection and management of AF, the underlying molecular mechanisms associated with the onset of atrial fibrillation and its progression remain still unclear. Among these molecular mechanisms, the implication of the adenosinergic system in AF has increased, since the accumulation of experimental data suggests that the increase in the adenosine blood level and the remodeling expression of the adenosine receptors might be part of the AF pathophysiology. Unfortunately, the adenosinergic system still has a Janus face in cardiac arrythmias, since adenosine can have both antiarrhythmic or proarrhythmic actions, along with adenosine receptors, which can lead to either profibrotic or antifibrotic effects.
- adenosine
- adenosine receptors
- atrial fibrillation
- arrhythmia
1. Adenosinergic System Signaling
1.1. Metabolism of Adenosine
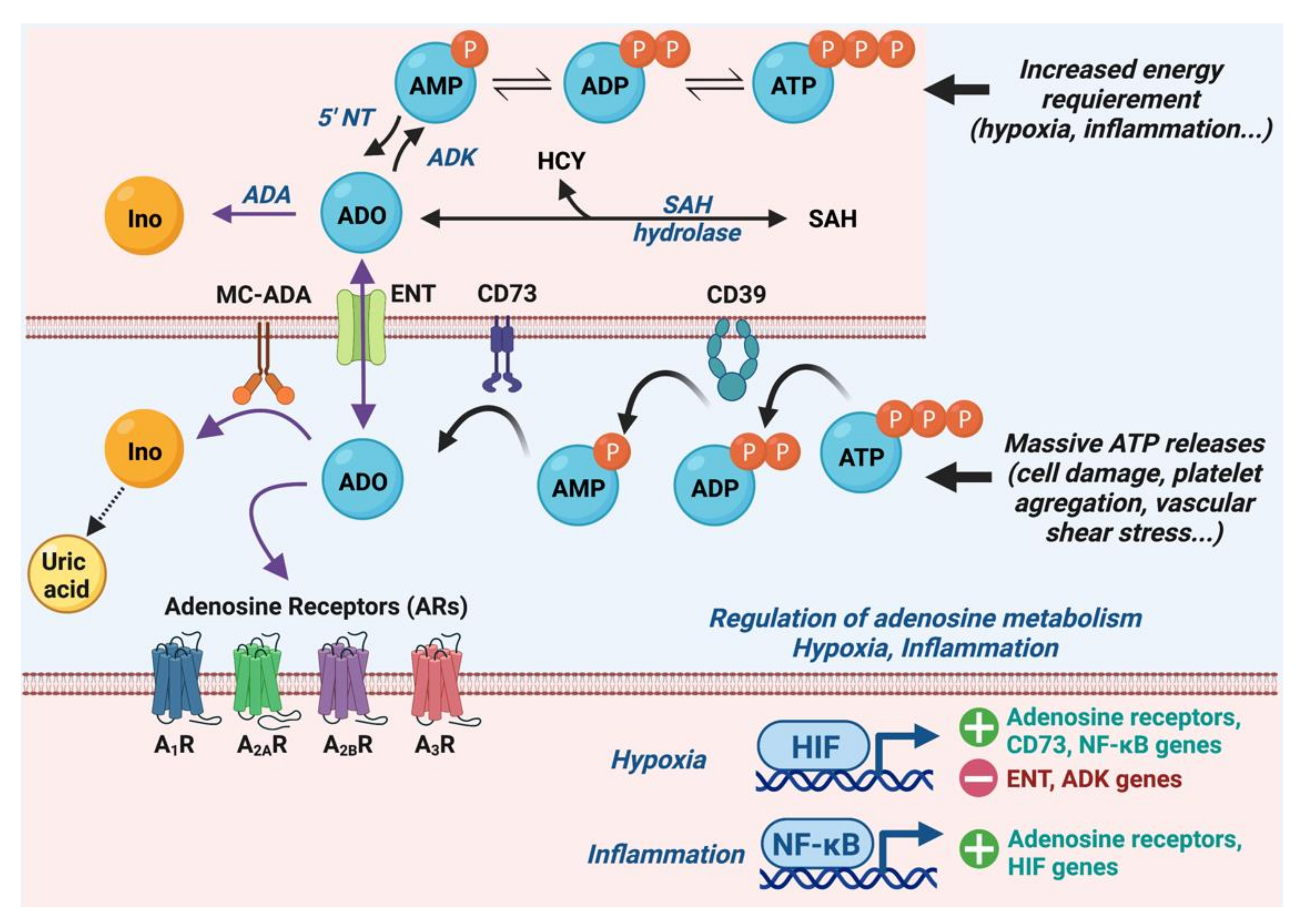
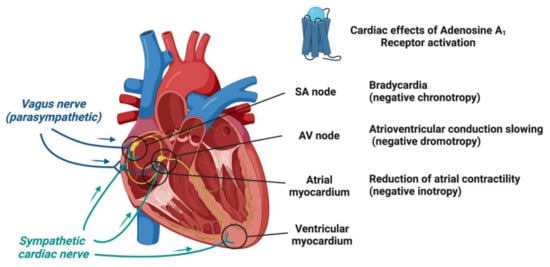
1.2. Adenosine Receptors and Their Cardiac Effects
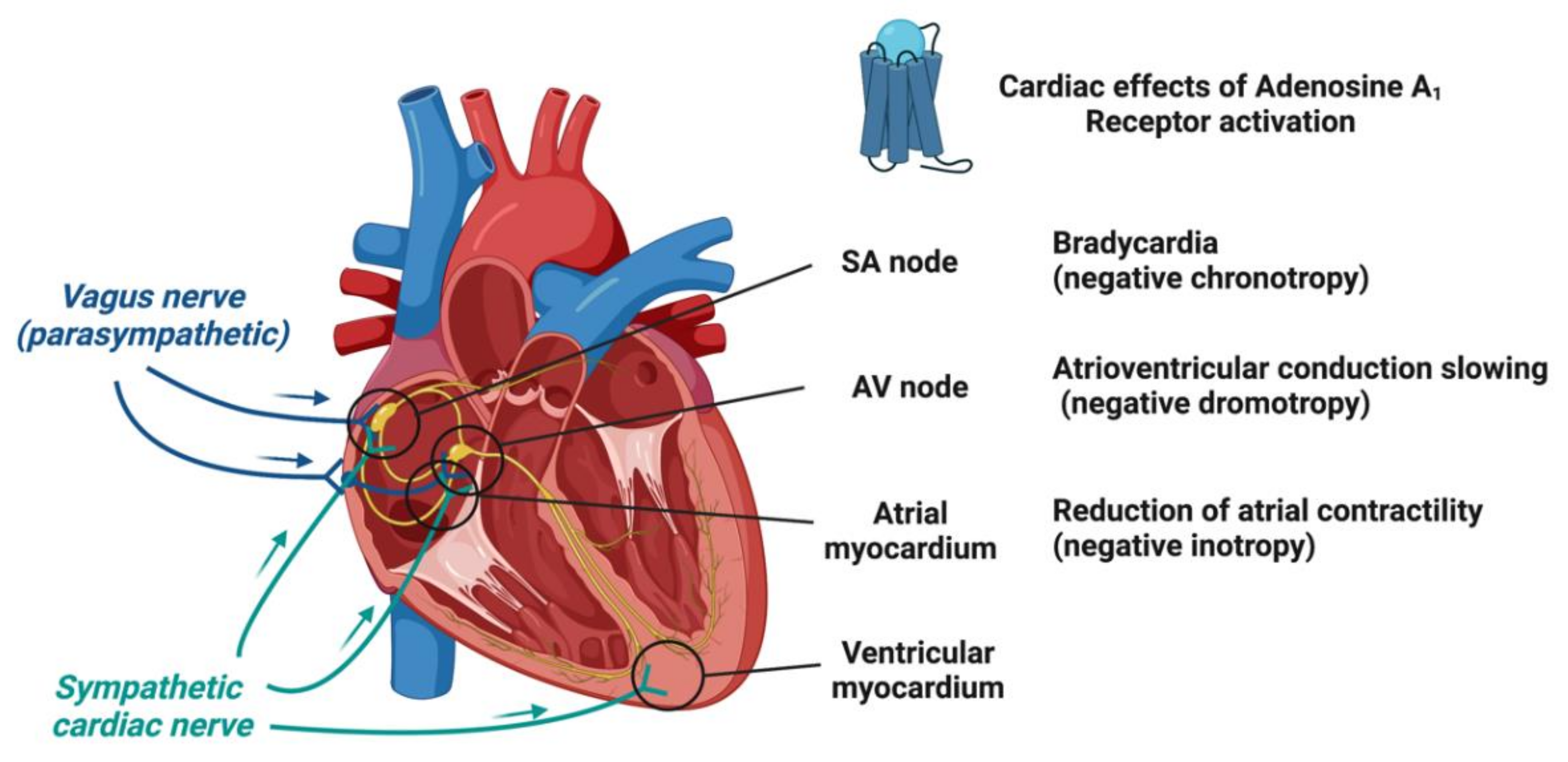
1.3. Molecular and Ionic Bases of Adenosine Effects in Cardiac Electrophysiology
The electrophysiological action of adenosine in the heart is regionally variable and dependent on the underlying ionic current population, which differ between species [21]. Irrespective of the species, adenosine exerts its cardiac electrophysiological actions mainly through the modulation of potassium, sodium and calcium currents by cardiac A1 and A2A receptors’ activation [22][23].1.3.1. Effects of Adenosine on Ionic Currents
Adenosine is believed to induce a cAMP-dependent (indirect effects) anti-β-adrenergic action by a dual mechanism: the activation of the presynaptic A1R limits the release of norepinephrine [24] and the activation of postsynaptic A1R blocks the effects of catecholamines by inhibiting adenylyl cyclase activity and then reducing intracellular cAMP levels. This indirect effect subsequently decreased the catecholamine-induced calcium inward current through L-type calcium channels (ICa,L) [25] and the sodium inward current (“funny” current (If) or pacemaker current) through hyperpolarization-activated cyclic nucleotide-gated (HCN) channels [26]. The activation of A1Rs enhances the IK,Ado thanks to G-protein-coupled, inwardly rectifying K+ (GIRK) channels [27]. After the Gi-protein activation, the Giα subunits enhance the gating of the GIRK channels, in the same manner as acetylcholine on the muscarinic-2 receptor, which causes a vagally mediated negative chronotropy upon on atrial pacemaker activity [27][28]. The GIRK channels aim to maintain the potassium equilibrium potential (EK+ = −90 mV) and modulate its conductance according to the membrane potential. As other inward rectifiers, they conduct larger inward currents when the membrane potential is negative to the EK+ than outward currents when the membrane potential is positive to EK+ [29][30][31]. On the contrary, like other GS-protein coupled receptors, A2AR stimulation can activate a cAMP-dependent pathway modulating Ca2+ handling. However, the precise mechanism of the A2AR-induced transduction signal remains controversial. Indeed, some authors report that the stimulation of A2ARs by a specific agonist (i.e., CGS 21680) did not alter the cAMP level in rat ventricular cardiomyocytes [32], whereas, more recently, others showed that CGS 21680 increased the cAMP content in ventricular cardiomyocytes of transgenic mice overexpressing human A2AR [33]. In the canonical Ca2+-dependent pathway, the PKA-dependent phosphorylation might increase the activation of L-type Ca2+ channels (LTCC) initiated by membrane depolarization. Following LTCC opening, the inward Ca2+ current (ICa,L), in turn, triggers a Ca2+-induced Ca2+ release (CIRC) from the sarcoplasmic reticulum via the phosphorylated cardiac ryanodine receptors (RyR2) and which is responsible for numerous spontaneous Ca2+ release events (Ca2+ spark and Ca2+ waves) during systole [34][35]. This can lead to the positive inotropic effect of A2AR stimulation in atria and ventricles through the Ca2+-induced myofibrilla contraction responsible for the cardiac electromechanical coupling [36]. The structural organization of cardiomyocytes into specific microdomains (i.e., cardiac dyads) favors this cardiac excitation–contraction coupling [37][38]. This is supported by the regional variation of the phosphorylation state of the LTCC [39] and the expression and the activity of the Ca2+ handling proteins [38][39][40]. Therefore, the effects of adenosine in the nodal cells (i.e., SAN and AVN), the conductive tissues and in the working myocardium are largely based on the modulation of adenylate cyclase activity, which induces cAMP-dependent PKA signaling, leading to Ca2+ handling protein phosphorylation, and to a GIRK-induced potassium outward current (IK,Ado) (Figure 3).
1.3.2. Adenosine Effects on the Action Potential in Nodal Cells and Working Cardiomyocytes
The action potential of the sinoatrial node cells is characterized by a more depolarized (−60 mV) and more labile resting membrane potential than contractile cardiomyocytes due to the almost lack of Kir2-encoded inwardly rectifier potassium currents (IK1) [41][42]. After a previous action potential, when the membrane potential reaches the maximum diastolic membrane potential, the HCN channels’ opening allows for the inward Na+ “funny” current (If), which contributes to the automaticity. This spontaneous diastolic HCN-mediated depolarization drives the membrane potential to the action potential threshold (~−40 mV) [43]. This threshold triggers the opening of voltage-dependent L-type Ca2+ channels, which induce a low slope of depolarization (phase 0). Lastly, while the Ca2+ channels close, the rapid and slow delayed rectifier current (IKr, IKs) induces outward K+ currents, which are responsible for the repolarization (Figure 4A).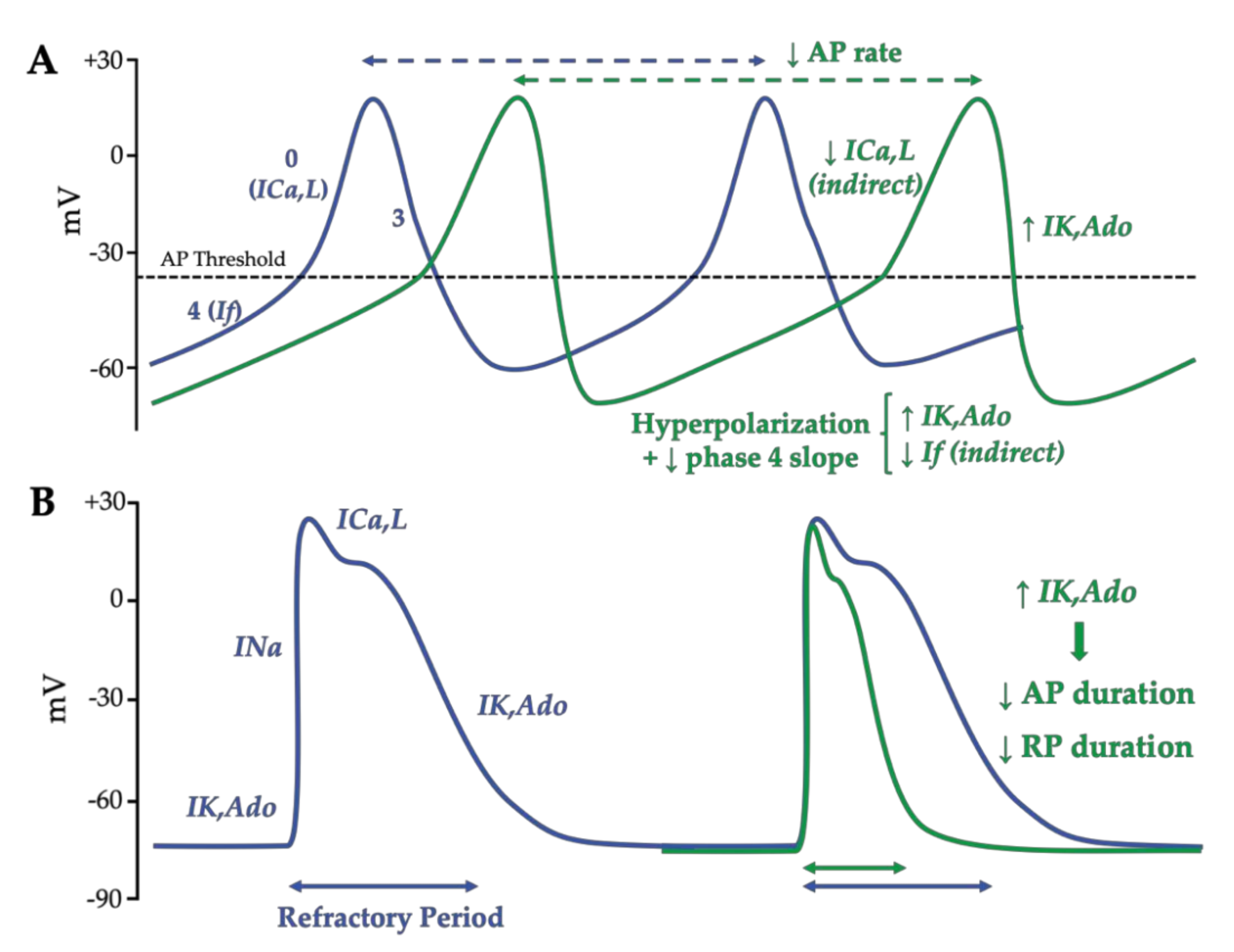
2. Pathophysiology of Atrial Fibrillation
2.1. Atrial Fibrillation Triggers
Repeated atrial ectopias are known to initiate AF and are considered as triggers of AF [47]. They can be localized within the pulmonary veins (PVs) and, less frequently, in others atrial areas (non-PV triggers). Among patients affected by AF, 20% of them exhibit non-PV foci [48], which can be predicted by female gender (odds ratio: 2.00), left atrial enlargement (odds ratio: 2.34) and AF episode prolongation [49][50]. Mapping studies have identified and localized a discrete clustered anatomical area corresponding to non-PV foci in the inferior mitral annulus, the posterior left atrium, the interatrial septum particularly at the fossa ovalis/limbus region, the crista terminalis and Eustachian ridge, the coronary sinus, and the superior vena cava [50]. PV ectopias originate all along the myocardium sleeve of PV and present unpredictable firing with various, intermittent and delayed conduction to the left atrium. They are defined as focal discrete sites of early and centrifugal activation [51]. Compared to the atrial cells, the PV cardiomyocytes have specific action potential properties that predispose to arrhythmogenesis [52]. Indeed, PV cells have a higher resting membrane potential, a lower amplitude of the action potential, a smaller maximum phase 0 upstroke velocity and a shorter action potential duration (Figure 5).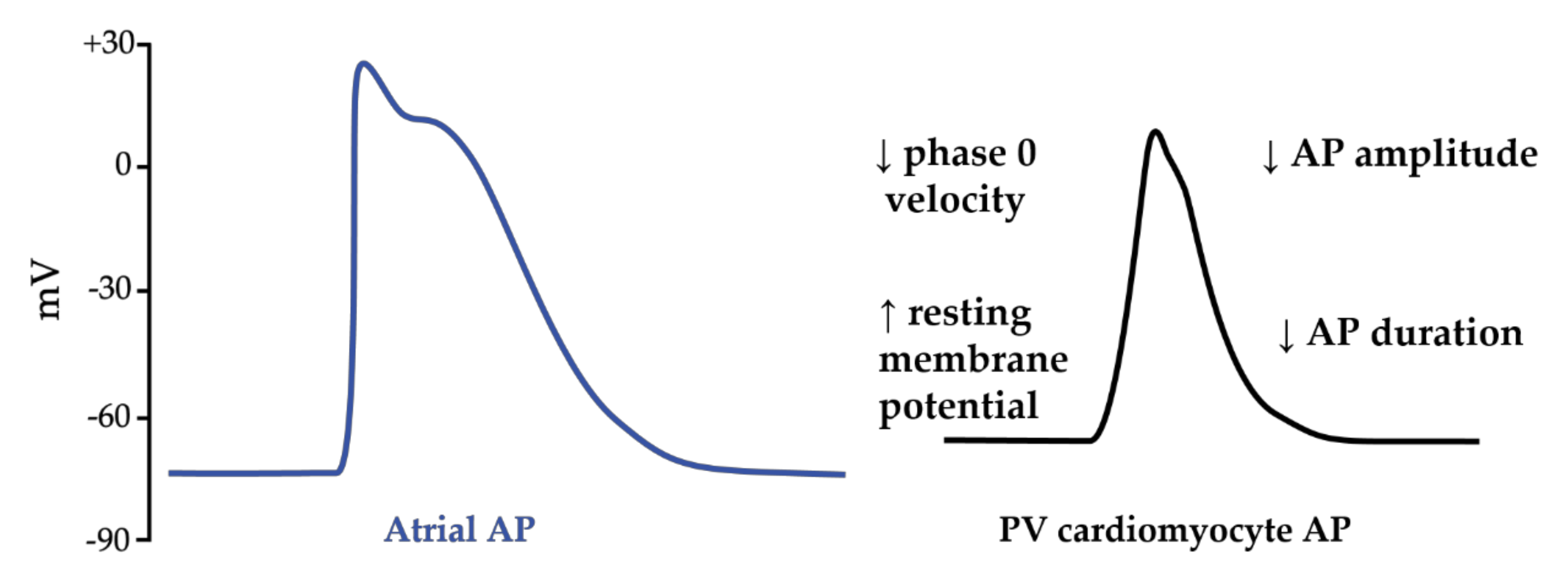
2.2. Mechanisms of AF Perpetuation
Two major mechanisms of perpetuation supported by anatomical and/or electrophysiological changes are described by the multiple wavelets hypothesis [46][53] and the localized (focal or reentrant) AF drivers. The multiple-wavelet phenomenon hypothesizes that multiple random wavefronts propagate through the atria until depolarizable tissue is available. Secondary to AF, atrial enlargement increases the critical atrial mass that is required to self-perpetuate this mechanism [54]. Within a same critical atrial mass, a short refractory period and delayed conduction velocities increase the theoretical number of wavelets. AF drivers are defined as a localized source of fast and repetitive activity during AF episodes from which activation propagates and breaks down into fibrillation of the rest of the atria [55]. The drivers are considered to be focal when the wavefront activation originates from a focal site with centrifugal activation and as reentrant when the waves fully and continuously rotate around an anatomical or functional pivot point [56]. As ectopias, because of the electrophysiological characteristics of PV cells and the brutal change in PV orientation fiber, the drivers are mostly located in the PV antral and adjacent regions [56][57]. With a longer AF duration, the complexity of the AF drivers increases, and they are located at extra-PV sites [56]. Anatomical reentries are defined by the presence of an unidirectional slow conduction area or block resulting in a fixed cycle length and localization circuit. Atrial fibrosis favors these slow conduction areas [58]. The nonuniform anisotropic conduction within a fibrosis area is an important substrate for reentrant tachycardia [53]. Functional reentries are defined by an absence of an underlying substrate and/or anatomical obstacle. Figure 6 illustrates the formation of a functional reentrant circuit due to the heterogeneity of the atrial potential duration at the PV junction. Then, the functional reentry can be schematized as a central refractory area maintained by centripetal waves moving around [59] or a propagation of spiral wave reentry or “rotor” around a core area at which the depolarization and the repolarization curves join each other [60].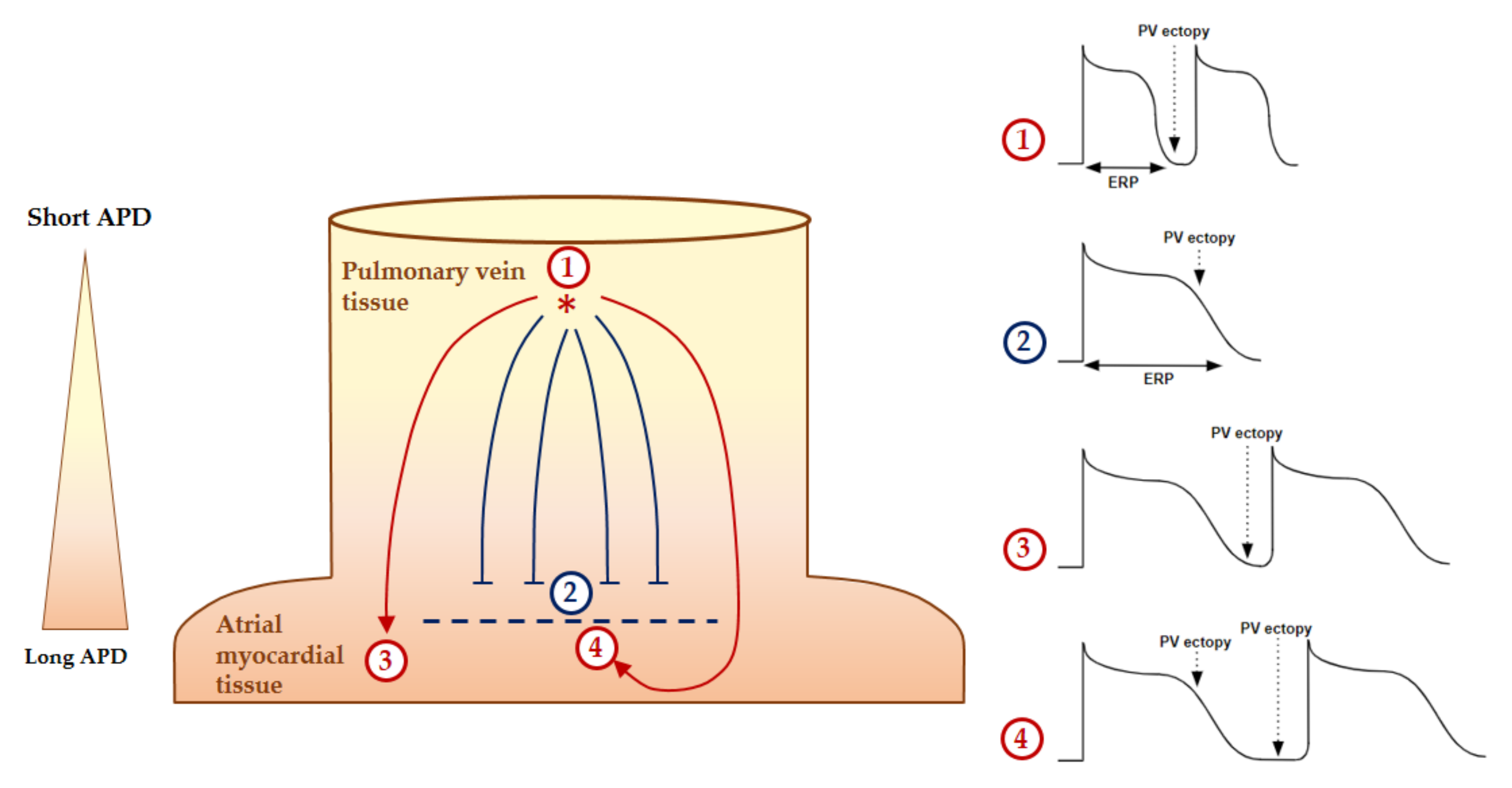
2.3. Substrate of Atrial Fibrillation and Atrial Cardiomyopathy
2.4. Pejorative Modulators of Atrial Fibrillation
3. Arrhythmogenic Effects of the Adenosinergic System
3.1. Adenosine Level and Expression of Adenosine Receptors in AF Patients
High adenosine plasma levels have been found in the left atria of patients during episodes of paroxysmal AF and in persistent AF [65]. The adenosine plasma concentrations then normalized after spontaneous or electrical cardioversion in sinus rhythm [65]. Moreover, the adenosine plasma concentrations in peripheric blood circulation were also higher in permanent AF compared to paroxysmal AF and controls [65]. The high adenosine plasma concentrations could be attributed to peripheral hypoxemia caused by the decrease in the left ventricular output in AF [65][66].
A high adenosine plasma concentration could also be a consequence of energy use in specific underlying cardiovascular conditions, including hypertension [67][68], chronic heart failure [69][70] or vagal syncope [71][72]. These are especially known to be AF risk factors. Interestingly, AF initiation has been described during the strong release of adenosine or the use of extrinsic adenosine injection [73][74].
3.2. Implication of A
1
Receptors in AF
In Langendorff preparation of rat hearts, the injection of a specific A1R agonist (CCPA) produced a profound negative chronotropic effect, whereas the selective A1R antagonist (PSB36) produced a nonsignificant positive chronotropic effect [75]. The increased concentration of both agonist and antagonist produced runs of repetitive atrial ectopy. This clinical effects appears to be mainly driven by a shortening of the action potential and effective refractory period durations, resulting in an increasing AF susceptibility due to the A1R activation [75]. Indeed, the activation of A1R through IK,Ado modulation induces a resting membrane potential hyperpolarization, a reduction of the action potential duration and a shortened effective refractory period (ERP) on atrial cardiomyocytes [27] (Figure 7).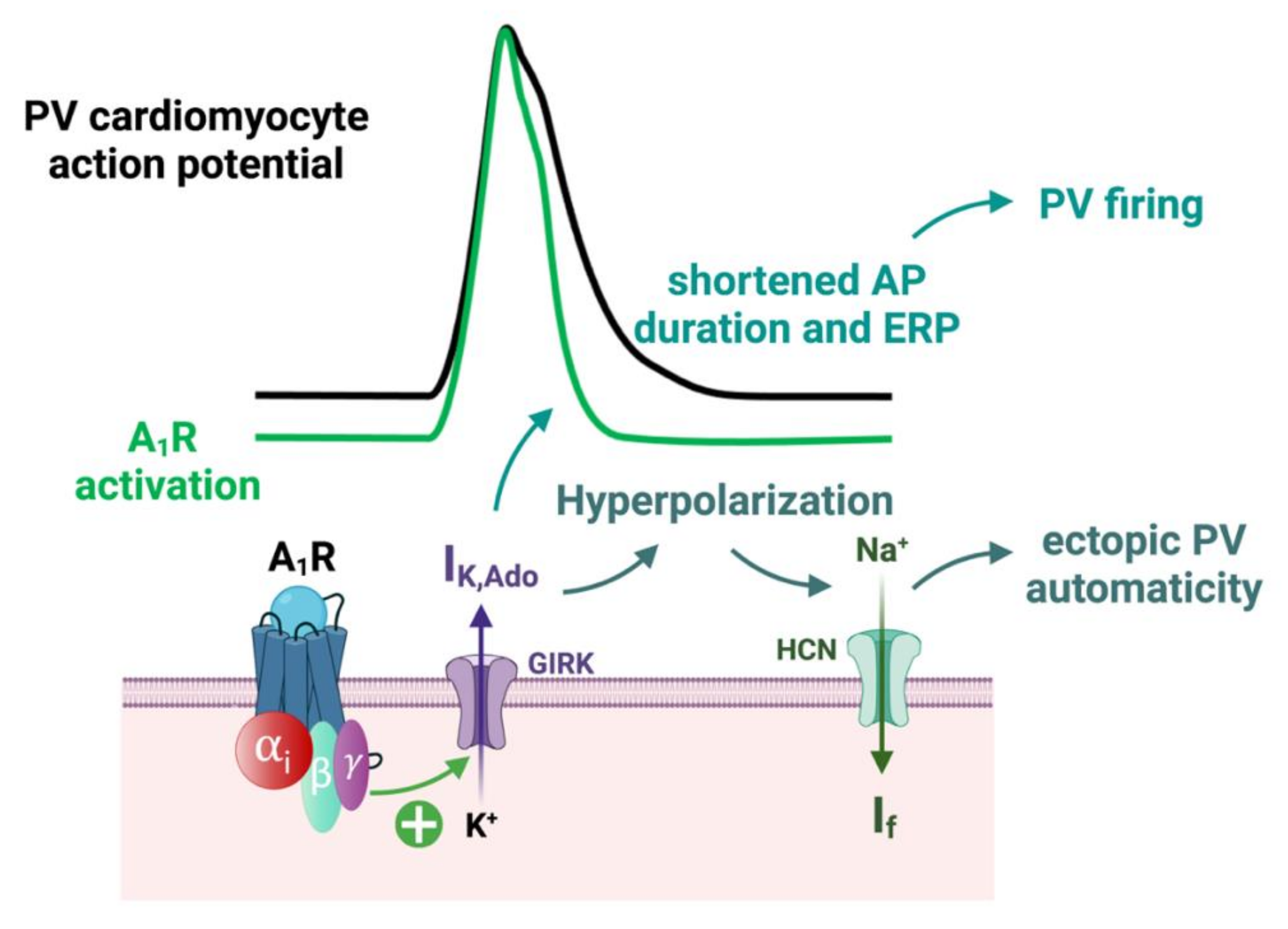
3.3. Implication of AR in the Remodeling of Calcium Handling
3.4. Modulation of Atrial Fibrosis by A
2B
Receptors
The activation of A2BR has an important role in cardiac fibroblast homeostasis, but the role of A2B in fibrosis is controversial, since both profibrotic [81][82][83][84] and antifibrotic effects are reported [85][86][87][88][89]. In cardiac fibroblasts, the increase in cAMP production following A2BR activation plays an essential role in the inhibition of angiotensin-II-induced collagen production [86]. However, activation of the A2BR increases the production of collagen and increases the release of IL-6 in human cardiac fibroblasts, resulting in a profibrosis state [84][90].4. Association between Atrial Fibrillation Risk Factors and the Adenosinergic System
Dysregulation of the autonomous nervous system are characterized by an excessive sympathetic activation, and a diminished parasympathetic influence is central to the pathogenesis of cardiovascular diseases, including heart failure, hypertension and AF [71]. Combined sympatho-vagal activation reflects the equilibrium between the release of epinephrine/norepinephrine, which activate the adrenergic receptors, and the release of acetylcholine, which induces the activation of the muscarinic receptors. Atrial sympathetic innervation is controlled by adrenergic receptors, which are divided into three major subfamilies: α1-, α2-, and β-adrenergic receptors. They are all coupled to different classes of heteromeric G proteins. The α1 and α2 receptors are coupled to Gq/11 and Gi/o, respectively. β-Adrenergic receptors are coupled to Gs. The α1 and α2 receptors induce a strong extracellular ATP release, while the β-adrenergic receptors activate cAMP production [7]. The activation of the β-adrenergic receptors by isoproterenol leads to the phosphorylation of nonjunctional RyR2 and L-type Ca2+ channels (LTCC) and is responsible for large increases in the Ca2+ flux [38]. Interestingly, after the use of an A2AR agonist, the increase in the heart rate was attenuated by a β-blocker. Thus, Wragg et al. demonstrated a direct activation of the sympathetic nervous system by A2AR stimulation [91]. In parallel, atrial parasympathetic innervation is controlled by muscarinic receptors (M2Rs). As with A1R stimulation, M2R activation leads to the opposite effect on β-adrenergic stimulation. The M2R stimulation activates inhibitory G proteins, subsequently reduces the activity of the HCN and LTCC and leads to decreased automaticity and conduction velocity in the nodal cell. M2R also activates the GIRK channel responsible for hyperpolarization [92]. Sympathovagal activation is a strong modulator of AF. Interestingly, hypertension, sleep apnea and heart failure are known to induce sympathetic tone activation and specific atrial remodeling [45][93][94][95][96]. Moreover, all three AF risk factors also induce specific adenosinergic system remodeling. Especially in patients suffering from essential hypertension, an A2AR overexpression was described in PBMCs [68]. Because of the vasodilator effect of A2AR, it was hypothesized that the A2AR overexpression was a compensatory mechanism of high blood pressure [96]. However, the chronic release of adenosine in the peripheral cardiovascular system during high blood pressure may also induce atrial remodeling of adenosine receptors. In the same manner, in sleep apnea or heart failure, the associated hypoxia may contribute to the atrial adenosinergic system’s remodeling and induce a pro-arrhythmogenic environment. As the A1R stimulation induces GIRK activation, the question remains whether A1R remodeling and its activation by adenosine can also contribute to arrhythmogenic effects through a similar pathway. The stimulation of the adrenergic and parasympathetic tone alone or combined can predispose to the AF onset [97]. Sequential combined stimulations had a synergic effect rather than vagal or sympathetic drive alone [97]. Interestingly, heart rate variability analyses before the occurrence of AF showed that AF patients have specific sympathetic and parasympathetic patterns [98]. Furthermore, prolonged atrial pacing in dogs induced sympathovagal activation and increased the risk of AF. Interestingly, the cryoablation of both autonomic nerves prevents the occurrence of AF [99]. However, the exact interaction between the adenosine receptors and cardiac autonomic innervation, as facilitator or inhibitor, is still unclear.References
- Drury, A.N.; Szent-Györgyi, A. The Physiological Activity of Adenine Compounds with Especial Reference to Their Action upon the Mammalian Heart. J. Physiol. 1929, 68, 213–237.
- Burnstock G Purinergic Nerves. Pharmacol. Rev. 1972, 24, 509–581.
- Szentmiklosi, A.J.; Galajda, Z.; Cseppento, Á.; Gesztelyi, R.; Susán, Z.; Hegyi, B.; Nánási, P.P. The Janus Face of Adenosine: Antiarrhythmic and Proarrhythmic Actions. Curr. Pharm. Des. 2015, 21, 965–976.
- Guieu, R.; Deharo, J.-C.; Maille, B.; Crotti, L.; Torresani, E.; Brignole, M.; Parati, G. Adenosine and the Cardiovascular System: The Good and the Bad. J. Clin. Med. 2020, 9, E1366.
- Fredholm, B.B. Adenosine--a Physiological or Pathophysiological Agent? J. Mol. Med. 2014, 92, 201–206.
- Deussen, A.; Lloyd, H.G.; Schrader, J. Contribution of S-Adenosylhomocysteine to Cardiac Adenosine Formation. J. Mol. Cell. Cardiol. 1989, 21, 773–782.
- Sumi, Y.; Woehrle, T.; Chen, Y.; Yao, Y.; Li, A.; Junger, W.G. Adrenergic Receptor Activation Involves ATP Release and Feedback through Purinergic Receptors. Am. J. Physiol. Cell Physiol. 2010, 299, C1118–C1126.
- Le, G.Y.; Essackjee, H.C.; Ballard, H.J. Intracellular Adenosine Formation and Release by Freshly-Isolated Vascular Endothelial Cells from Rat Skeletal Muscle: Effects of Hypoxia and/or Acidosis. Biochem. Biophys. Res. Commun. 2014, 450, 93–98.
- Colgan, S.P.; Eltzschig, H.K.; Eckle, T.; Thompson, L.F. Physiological Roles for Ecto-5’-Nucleotidase (CD73). Purinerg. Signal. 2006, 2, 351–360.
- Fredholm, B.B.; Arslan, G.; Halldner, L.; Kull, B.; Schulte, G.; Wasserman, W. Structure and Function of Adenosine Receptors and Their Genes. Naunyn. Schmiedebergs. Arch. Pharmacol. 2000, 362, 364–374.
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and Classification of Adenosine Receptors. Pharmacol. Rev. 2001, 53, 527–552.
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625.
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228.
- Headrick, J.P.; Ashton, K.J.; Rose’meyer, R.B.; Peart, J.N. Cardiovascular Adenosine Receptors: Expression, Actions and Interactions. Pharmacol. Ther. 2013, 140, 92–111.
- Chandrasekera, P.C.; McIntosh, V.J.; Cao, F.X.; Lasley, R.D. Differential Effects of Adenosine A2a and A2b Receptors on Cardiac Contractility. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H2082–H2089.
- Musser, B.; Morgan, M.E.; Leid, M.; Murray, T.F.; Linden, J.; Vestal, R.E. Species Comparison of Adenosine and Beta-Adrenoceptors in Mammalian Atrial and Ventricular Myocardium. Eur. J. Pharmacol. 1993, 246, 105–111.
- Shryock, J.C.; Belardinelli, L. Adenosine and Adenosine Receptors in the Cardiovascular System: Biochemistry, Physiology, and Pharmacology. Am. J. Cardiol. 1997, 79, 2–10.
- Schrader, J.; Baumann, G.; Gerlach, E. Adenosine as Inhibitor of Myocardial Effects of Catecholamines. Pflugers. Arch. 1977, 372, 29–35.
- Dobson, J.G. Mechanism of Adenosine Inhibition of Catecholamine-Induced Responses in Heart. Circ. Res. 1983, 52, 151–160.
- Wennmalm, M.; Fredholm, B.B.; Hedqvist, P. Adenosine as a Modulator of Sympathetic Nerve-Stimulation-Induced Release of Noradrenaline from the Isolated Rabbit Heart. Acta. Physiol. Scand. 1988, 132, 487–494.
- Belardinelli, L.; Giles, W.R.; West, A. Ionic Mechanisms of Adenosine Actions in Pacemaker Cells from Rabbit Heart. J. Physiol. 1988, 405, 615–633.
- Lerman, B.B.; Markowitz, S.M.; Cheung, J.W.; Liu, C.F.; Thomas, G.; Ip, J.E. Supraventricular Tachycardia. Circ. Arrhythmia Elec. 2018, 11, e006953.
- Brugada, J.; Katritsis, D.G.; Arbelo, E.; Arribas, F.; Bax, J.J.; Blomström-Lundqvist, C.; Calkins, H.; Corrado, D.; Deftereos, S.G.; Diller, G.-P.; et al. 2019 ESC Guidelines for the Management of Patients with Supraventricular TachycardiaThe Task Force for the Management of Patients with Supraventricular Tachycardia of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 41, 655–720.
- Lokhandwala, M.F. Inhibition of Cardiac Sympathetic Neurotransmission by Adenosine. Eur. J. Pharmacol. 1979, 60, 353–357.
- Belardinelli, L.; Shryock, J.C.; Song, Y.; Wang, D.; Srinivas, M. Ionic Basis of the Electrophysiological Actions of Adenosine on Cardiomyocytes. FASEB J. 1995, 9, 359–365.
- DiFrancesco, D.; Tortora, P. Direct Activation of Cardiac Pacemaker Channels by Intracellular Cyclic AMP. Nature 1991, 351, 145–147.
- Wang, X.; Liang, B.; Skibsbye, L.; Olesen, S.-P.; Grunnet, M.; Jespersen, T. GIRK Channel Activation via Adenosine or Muscarinic Receptors Has Similar Effects on Rat Atrial Electrophysiology. J. Cardiovasc. Pharmacol. 2013, 62, 192–198.
- Pfaffinger, P.J.; Martin, J.M.; Hunter, D.D.; Nathanson, N.M.; Hille, B. GTP-Binding Proteins Couple Cardiac Muscarinic Receptors to a K Channel. Nature 1985, 317, 536–538.
- Lopatin, A.N.; Nichols, C.G. Inward Rectifiers in the Heart: An Update on I(K1). J. Mol. Cell. Cardiol. 2001, 33, 625–638.
- Anumonwo, J.M.B.; Lopatin, A.N. Cardiac Strong Inward Rectifier Potassium Channels. J. Mol. Cell. Cardiol. 2010, 48, 45–54.
- Guo, D.; Ramu, Y.; Klem, A.M.; Lu, Z. Mechanism of Rectification in Inward-Rectifier K+ Channels. J. Gen. Physiol. 2003, 121, 261–276.
- Kilpatrick, E.L.; Narayan, P.; Mentzer, R.M.; Lasley, R.D. Cardiac Myocyte Adenosine A2a Receptor Activation Fails to Alter CAMP or Contractility: Role of Receptor Localization. Am. J. Physiol. Heart. Circ. Physiol. 2002, 282, H1035–H1040.
- Boknik, P.; Drzewiecki, K.; Eskandar, J.; Gergs, U.; Grote-Wessels, S.; Fabritz, L.; Kirchhof, P.; Müller, F.U.; Stümpel, F.; Schmitz, W.; et al. Phenotyping of Mice with Heart Specific Overexpression of A2A-Adenosine Receptors: Evidence for Cardioprotective Effects of A2A-Adenosine Receptors. Front. Pharmacol. 2018, 9, 13.
- Hove-Madsen, L.; Prat-Vidal, C.; Llach, A.; Ciruela, F.; Casadó, V.; Lluis, C.; Bayes-Genis, A.; Cinca, J.; Franco, R. Adenosine A2A Receptors Are Expressed in Human Atrial Myocytes and Modulate Spontaneous Sarcoplasmic Reticulum Calcium Release. Cardiovasc. Res. 2006, 72, 292–302.
- Llach, A.; Molina, C.E.; Prat-Vidal, C.; Fernandes, J.; Casadó, V.; Ciruela, F.; Lluís, C.; Franco, R.; Cinca, J.; Hove-Madsen, L. Abnormal calcium handling in atrial fibrillation is linked to up-regulation of adenosine A2A receptors. Eur. Heart J. 2011, 32, 721–729.
- Boknik, P.; Eskandar, J.; Hofmann, B.; Zimmermann, N.; Neumann, J.; Gergs, U. Role of Cardiac A2A Receptors Under Normal and Pathophysiological Conditions. Front. Pharmacol. 2020, 11, 627838.
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195.
- Brandenburg, S.; Pawlowitz, J.; Steckmeister, V.; Subramanian, H.; Uhlenkamp, D.; Scardigli, M.; Mushtaq, M.; Amlaz, S.I.; Kohl, T.; Wegener, J.W.; et al. A Junctional CAMP Compartment Regulates Rapid Ca2+ Signaling in Atrial Myocytes. J. Mol. Cell. Cardiol. 2022, 165, 141–157.
- Christ, T.; Boknik, P.; Wöhrl, S.; Wettwer, E.; Graf, E.M.; Bosch, R.F.; Knaut, M.; Schmitz, W.; Ravens, U.; Dobrev, D. L-Type Ca2+ Current Downregulation in Chronic Human Atrial Fibrillation Is Associated With Increased Activity of Protein Phosphatases. Circulation 2004, 110, 2651–2657.
- Bokník, P.; Unkel, C.; Kirchhefer, U.; Kleideiter, U.; Klein-Wiele, O.; Knapp, J.; Linck, B.; Lüss, H.; Ulrich Müller, F.; Schmitz, W.; et al. Regional Expression of Phospholamban in the Human Heart. Cardiovasc. Res. 1999, 43, 67–76.
- Dhamoon, A.S.; Pandit, S.V.; Sarmast, F.; Parisian, K.R.; Guha, P.; Li, Y.; Bagwe, S.; Taffet, S.M.; Anumonwo, J.M.B. Unique Kir2.x Properties Determine Regional and Species Differences in the Cardiac Inward Rectifier K+ Current. Circ. Res. 2004, 94, 1332–1339.
- Chandler, N.J.; Greener, I.D.; Tellez, J.O.; Inada, S.; Musa, H.; Molenaar, P.; Difrancesco, D.; Baruscotti, M.; Longhi, R.; Anderson, R.H.; et al. Molecular Architecture of the Human Sinus Node: Insights into the Function of the Cardiac Pacemaker. Circulation 2009, 119, 1562–1575.
- MacDonald, E.A.; Rose, R.A.; Quinn, T.A. Neurohumoral Control of Sinoatrial Node Activity and Heart Rate: Insight From Experimental Models and Findings From Humans. Front. Physiol. 2020, 11, 170.
- Kléber, A.G.; Rudy, Y. Basic Mechanisms of Cardiac Impulse Propagation and Associated Arrhythmias. Physiol. Rev. 2004, 84, 431–488.
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the Diagnosis and Management of Atrial Fibrillation Developed in Collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2021, 42, 373–498.
- Cheniti, G.; Vlachos, K.; Pambrun, T.; Hooks, D.; Frontera, A.; Takigawa, M.; Bourier, F.; Kitamura, T.; Lam, A.; Martin, C.; et al. Atrial Fibrillation Mechanisms and Implications for Catheter Ablation. Front. Physiol. 2018, 9, 1458.
- Haïssaguerre, M.; Jaïs, P.; Shah, D.C.; Takahashi, A.; Hocini, M.; Quiniou, G.; Garrigue, S.; Le Mouroux, A.; Le Métayer, P.; Clémenty, J. Spontaneous Initiation of Atrial Fibrillation by Ectopic Beats Originating in the Pulmonary Veins. N. Engl. J. Med. 1998, 339, 659–666.
- Lin, W.-S.; Tai, C.-T.; Hsieh, M.-H.; Tsai, C.-F.; Lin, Y.-K.; Tsao, H.-M.; Huang, J.-L.; Yu, W.-C.; Yang, S.-P.; Ding, Y.-A.; et al. Catheter Ablation of Paroxysmal Atrial Fibrillation Initiated by Non–Pulmonary Vein Ectopy. Circulation 2003, 107, 3176–3183.
- Lee, S.-H.; Tai, C.-T.; Hsieh, M.-H.; Tsao, H.-M.; Lin, Y.-J.; Chang, S.-L.; Huang, J.-L.; Lee, K.-T.; Chen, Y.-J.; Cheng, J.-J.; et al. Predictors of Non-Pulmonary Vein Ectopic Beats Initiating Paroxysmal Atrial Fibrillation: Implication for Catheter Ablation. J. Am. Coll. Cardiol. 2005, 46, 1054–1059.
- Santangeli, P.; Marchlinski, F.E. Techniques for the Provocation, Localization, and Ablation of Non–Pulmonary Vein Triggers for Atrial Fibrillation. Heart Rhythm 2017, 14, 1087–1096.
- Arentz, T.; Haegeli, L.; Sanders, P.; Weber, R.; Neumann, F.J.; Kalusche, D.; Haïssaguerre, M. High-Density Mapping of Spontaneous Pulmonary Vein Activity Initiating Atrial Fibrillation in Humans. J. Cardiovasc. Electrophysiol. 2007, 18, 31–38.
- Ehrlich, J.R.; Cha, T.-J.; Zhang, L.; Chartier, D.; Melnyk, P.; Hohnloser, S.H.; Nattel, S. Cellular Electrophysiology of Canine Pulmonary Vein Cardiomyocytes: Action Potential and Ionic Current Properties. J. Physiol. 2003, 551, 801–813.
- Haissaguerre, M.; Shah, A.J.; Cochet, H.; Hocini, M.; Dubois, R.; Efimov, I.; Vigmond, E.; Bernus, O.; Trayanova, N. Intermittent Drivers Anchoring to Structural Heterogeneities as a Major Pathophysiological Mechanism of Human Persistent Atrial Fibrillation. J. Physiol. 2016, 594, 2387–2398.
- Qu, Z. Critical Mass Hypothesis Revisited: Role of Dynamical Wave Stability in Spontaneous Termination of Cardiac Fibrillation. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H255–H263.
- Hansen, B.J.; Csepe, T.A.; Zhao, J.; Ignozzi, A.J.; Hummel, J.D.; Fedorov, V.V. Maintenance of Atrial Fibrillation: Are Reentrant Drivers With Spatial Stability the Key? Circ. Arrhythm. Electrophysiol. 2016, 9, e004398.
- Lim, H.S.; Hocini, M.; Dubois, R.; Denis, A.; Derval, N.; Zellerhoff, S.; Yamashita, S.; Berte, B.; Mahida, S.; Komatsu, Y.; et al. Complexity and Distribution of Drivers in Relation to Duration of Persistent Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 69, 1257–1269.
- Roney, C.H.; Bayer, J.D.; Cochet, H.; Meo, M.; Dubois, R.; Jaïs, P.; Vigmond, E.J. Variability in Pulmonary Vein Electrophysiology and Fibrosis Determines Arrhythmia Susceptibility and Dynamics. PLOS Comput. Biol. 2018, 14, e1006166.
- Frontera, A.; Pagani, S.; Limite, L.R.; Peirone, A.; Fioravanti, F.; Enache, B.; Cuellar, S.J.; Vlachos, K.; Meyer, C.; Montesano, G.; et al. Slow Conduction Corridors and Pivot Sites Characterize the Electrical Remodeling in Atrial Fibrillation. JACC Cli. Electrophysiol. 2022, 8, 561–577.
- Allessie, M.A.; Bonke, F.I.; Schopman, F.J. Circus Movement in Rabbit Atrial Muscle as a Mechanism of Tachycardia. III. The “Leading Circle” Concept: A New Model of Circus Movement in Cardiac Tissue without the Involvement of an Anatomical Obstacle. Circ. Res. 1977, 41, 9–18.
- Nattel, S.; Xiong, F.; Aguilar, M. Demystifying Rotors and Their Place in Clinical Translation of Atrial Fibrillation Mechanisms. Nat. Rev. Cardiol. 2017, 14, 509–520.
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological Mechanisms of Atrial Fibrillation: A Translational Appraisal. Physiol. Rev. 2011, 91, 265–325.
- Maille, B.; Das, M.; Hussein, A.; Shaw, M.; Chaturvedi, V.; Williams, E.; Morgan, M.; Ronayne, C.; Snowdon, R.L.; Gupta, D. Reverse Electrical and Structural Remodeling of the Left Atrium Occurs Early after Pulmonary Vein Isolation for Persistent Atrial Fibrillation. J. Interv. Card. Electrophysiol. 2020, 58, 9–19.
- Goette, A.; Kalman, J.M.; Aguinaga, L.; Akar, J.; Cabrera, J.A.; Chen, S.A.; Chugh, S.S.; Corradi, D.; D’Avila, A.; Dobrev, D.; et al. EHRA/HRS/APHRS/SOLAECE Expert Consensus on Atrial Cardiomyopathies: Definition, Characterization, and Clinical Implication. EP Europace 2016, 18, 1455–1490.
- Heijman, J.; Guichard, J.-B.; Dobrev, D.; Nattel, S. Translational Challenges in Atrial Fibrillation. Circ. Res. 2018, 122, 752–773.
- Maille, B.; Marlinge, M.; Vairo, D.; Mottola, G.; Koutbi, L.; Deharo, P.; Gastaldi, M.; Gaudry, M.; Guiol, C.; Bottone, S.; et al. Adenosine Plasma Level in Patients with Paroxysmal or Persistent Atrial Fibrillation and Normal Heart during Ablation Procedure and/or Cardioversion. Purinergic Signal. 2019, 15, 45–52.
- Cha, Y.-M.; Dzeja, P.P.; Shen, W.K.; Jahangir, A.; Hart, C.Y.T.; Terzic, A.; Redfield, M.M. Failing Atrial Myocardium: Energetic Deficits Accompany Structural Remodeling and Electrical Instability. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1313–H1320.
- Zhang, W.; Zhang, Y.; Wang, W.; Dai, Y.; Ning, C.; Luo, R.; Sun, K.; Glover, L.; Grenz, A.; Sun, H.; et al. Elevated Ecto-5’-Nucleotidase-Mediated Increased Renal Adenosine Signaling via A2B Adenosine Receptor Contributes to Chronic Hypertension. Circ. Res. 2013, 112, 1466–1478.
- Silhol, F.; Marlinge, M.; Guiol, C.; Chefrour, M.; Mace, P.; Criado, C.; Kipson, N.; Vaisse, B.; Vairo, D.; Sarlon, G.; et al. Characterization of Adenosine A2 Receptors in Peripheral Blood Mononuclear Cells of Patients with Fibromuscular Dysplasia. Hypertens. Res. 2020, 43, 466–469.
- Gaubert, M.; Marlinge, M.; Kerbaul, F.; Resseguier, N.; Laine, M.; Cautella, J.; Cordier, C.; Colomb, B.; Kipson, N.; Thuny, F.; et al. Adenosine Plasma Level and A2A Receptor Expression in Patients With Cardiogenic Shock. Crit. Care Med. 2018, 46, e874–e880.
- Franceschi, F.; Deharo, J.-C.; Giorgi, R.; By, Y.; Monserrat, C.; Condo, J.; Ibrahim, Z.; Saadjian, A.; Guieu, R. Peripheral Plasma Adenosine Release in Patients with Chronic Heart Failure. Heart 2009, 95, 651–655.
- Linz, D.; Elliott, A.D.; Hohl, M.; Malik, V.; Schotten, U.; Dobrev, D.; Nattel, S.; Böhm, M.; Floras, J.; Lau, D.H.; et al. Role of Autonomic Nervous System in Atrial Fibrillation. Int. J. Cardiol. 2019, 287, 181–188.
- Saadjian, A.Y.; Lévy, S.; Franceschi, F.; Zouher, I.; Paganelli, F.; Guieu, R.P. Role of Endogenous Adenosine as a Modulator of Syncope Induced during Tilt Testing. Circulation 2002, 106, 569–574.
- Tebbenjohanns, J.; Schumacher, B.; Pfeiffer, D.; Jung, W.; Lüderitz, B. Dose and Rate-Dependent Effects of Adenosine on Atrial Action Potential Duration in Humans. J. Interv. Card. Electrophysiol. 1997, 1, 33–37.
- Strickberger, S.A.; Man, K.C.; Daoud, E.G.; Goyal, R.; Brinkman, K.; Knight, B.P.; Weiss, R.; Bahu, M.; Morady, F. Adenosine-Induced Atrial Arrhythmia: A Prospective Analysis. Ann. Intern. Med. 1997, 127, 417–422.
- Soattin, L.; Lubberding, A.F.; Bentzen, B.H.; Christ, T.; Jespersen, T. Inhibition of Adenosine Pathway Alters Atrial Electrophysiology and Prevents Atrial Fibrillation. Front. Physiol. 2020, 11, 493.
- Van Wagoner, D.R.; Pond, A.L.; Lamorgese, M.; Rossie, S.S.; McCarthy, P.M.; Nerbonne, J.M. Atrial L-Type Ca2+ Currents and Human Atrial Fibrillation. Circ. Res. 1999, 85, 428–436.
- Reinhardt, F.; Beneke, K.; Pavlidou, N.G.; Conradi, L.; Reichenspurner, H.; Hove-Madsen, L.; Molina, C.E. Abnormal Calcium Handling in Atrial Fibrillation Is Linked to Changes in Cyclic AMP Dependent Signaling. Cells 2021, 10, 3042.
- Lehnart, S.E.; Wehrens, X.H.T.; Laitinen, P.J.; Reiken, S.R.; Deng, S.-X.; Cheng, Z.; Landry, D.W.; Kontula, K.; Swan, H.; Marks, A.R. Sudden Death in Familial Polymorphic Ventricular Tachycardia Associated with Calcium Release Channel (Ryanodine Receptor). Leak. Circulation 2004, 109, 3208–3214.
- Burashnikov, A.; Antzelevitch, C. Reinduction of Atrial Fibrillation Immediately after Termination of the Arrhythmia Is Mediated by Late Phase 3 Early Afterdepolarization-Induced Triggered Activity. Circulation 2003, 107, 2355–2360.
- Visentin, S.; Wu, S.N.; Belardinelli, L. Adenosine-Induced Changes in Atrial Action Potential: Contribution of Ca and K Currents. Am. J. Physiol. 1990, 258, H1070–H1078.
- Vecchio, E.A.; White, P.J.; May, L.T. Targeting Adenosine Receptors for the Treatment of Cardiac Fibrosis. Front. Pharmacol. 2017, 8, 243.
- Vecchio, E.A.; White, P.J.; May, L.T. The Adenosine A2B G Protein-Coupled Receptor: Recent Advances and Therapeutic Implications. Pharmacol. Ther. 2019, 198, 20–33.
- Toldo, S.; Zhong, H.; Mezzaroma, E.; Van Tassell, B.W.; Kannan, H.; Zeng, D.; Belardinelli, L.; Voelkel, N.F.; Abbate, A. GS-6201, a Selective Blocker of the A2B Adenosine Receptor, Attenuates Cardiac Remodeling after Acute Myocardial Infarction in the Mouse. J. Pharmacol. Exp. Ther. 2012, 343, 587–595.
- Zhong, H.; Belardinelli, L.; Zeng, D. Pro-Fibrotic Role of the A2B Adenosine Receptor in Human Cardiac Fibroblasts. J. Card. Fail. 2011, 17, S65.
- Lu, D.; Insel, P.A. Hydrolysis of Extracellular ATP by Ectonucleoside Triphosphate Diphosphohydrolase (ENTPD) Establishes the Set Point for Fibrotic Activity of Cardiac Fibroblasts. J. Biol. Chem. 2013, 288, 19040–19049.
- Phosri, S.; Bunrukchai, K.; Parichatikanond, W.; Sato, V.H.; Mangmool, S. Epac Is Required for Exogenous and Endogenous Stimulation of Adenosine A2B Receptor for Inhibition of Angiotensin II-Induced Collagen Synthesis and Myofibroblast Differentiation. Purinergic Signal. 2018, 14, 141–156.
- Chen, Y.; Epperson, S.; Makhsudova, L.; Ito, B.; Suarez, J.; Dillmann, W.; Villarreal, F. Functional Effects of Enhancing or Silencing Adenosine A2b Receptors in Cardiac Fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2478–H2486.
- Wakeno, M.; Minamino, T.; Seguchi, O.; Okazaki, H.; Tsukamoto, O.; Okada, K.; Hirata, A.; Fujita, M.; Asanuma, H.; Kim, J.; et al. Long-Term Stimulation of Adenosine A2b Receptors Begun After Myocardial Infarction Prevents Cardiac Remodeling in Rats. Circulation 2006, 114, 1923–1932.
- Maas, J.E.; Wan, T.C.; Figler, R.A.; Gross, G.J.; Auchampach, J.A. Evidence That the Acute Phase of Ischemic Preconditioning Does Not Require Signaling by the A2B Adenosine Receptor. J. Mol. Cell. Cardiol. 2010, 49, 886–893.
- Feng, W.; Song, Y.; Chen, C.; Lu, Z.Z.; Zhang, Y. Stimulation of Adenosine A2B Receptors Induces Interleukin-6 Secretion in Cardiac Fibroblasts via the PKC-Delta-P38 Signalling Pathway. Br. J. Pharmacol. 2010, 159, 1598–1607.
- Wragg, E.S.; Pannucci, P.; Hill, S.J.; Woolard, J.; Cooper, S.L. Involvement of β-Adrenoceptors in the Cardiovascular Responses Induced by Selective Adenosine A2A and A2B Receptor Agonists. Pharmacol. Res. Perspect. 2022, 10, e00975.
- Lee, S.W.; Anderson, A.; Guzman, P.A.; Nakano, A.; Tolkacheva, E.G.; Wickman, K. Atrial GIRK Channels Mediate the Effects of Vagus Nerve Stimulation on Heart Rate Dynamics and Arrhythmogenesis. Front. Physiol. 2018, 9, 943.
- Javed, S.; Gupta, D.; Lip, G.Y.H. Obesity and Atrial Fibrillation: Making Inroads through Fat. Eur. Heart J. Cardiovasc. Pharmacother. 2021, 7, 59–67.
- Nalliah, C.J.; Wong, G.R.; Lee, G.; Voskoboinik, A.; Kee, K.; Goldin, J.; Watts, T.; Linz, D.; Wirth, D.; Parameswaran, R.; et al. Sleep Apnoea Has a Dose-Dependent Effect on Atrial Remodelling in Paroxysmal but Not Persistent Atrial Fibrillation: A High-Density Mapping Study. EP Europace 2021, 23, 691–700.
- Sidhu, K.; Tang, A. Modifiable Risk Factors in Atrial Fibrillation: The Role of Alcohol, Obesity, and Sleep Apnea. Can. J. Cardiol. 2017, 33, 947–949.
- Sheng, Y.; Li, M.; Xu, M.; Zhang, Y.; Xu, J.; Huang, Y.; Li, X.; Yao, G.; Sui, W.; Zhang, M.; et al. Left Ventricular and Atrial Remodelling in Hypertensive Patients Using Thresholds from International Guidelines and EMINCA Data. Eur. Heart J. Cardiovasc. Imag. 2022, 23, 166–174.
- Sharifov, O.F.; Fedorov, V.V.; Beloshapko, G.G.; Glukhov, A.V.; Yushmanova, A.V.; Rosenshtraukh, L.V. Roles of Adrenergic and Cholinergic Stimulation in Spontaneous Atrial Fibrillation in Dogs. J. Am. Coll. Cardiol. 2004, 43, 483–490.
- Huang, J.L.; Wen, Z.C.; Lee, W.L.; Chang, M.S.; Chen, S.A. Changes of Autonomic Tone before the Onset of Paroxysmal Atrial Fibrillation. Int. J. Cardiol. 1998, 66, 275–283.
- Tan, A.Y.; Zhou, S.; Ogawa, M.; Song, J.; Chu, M.; Li, H.; Fishbein, M.C.; Lin, S.-F.; Chen, L.S.; Chen, P.-S. Neural Mechanisms of Paroxysmal Atrial Fibrillation and Paroxysmal Atrial Tachycardia in Ambulatory Canines. Circulation 2008, 118, 916–925.