Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Margaret Ho | -- | 2879 | 2022-11-30 05:41:41 | | | |
| 2 | Conner Chen | Meta information modification | 2879 | 2022-11-30 12:59:41 | | | | |
| 3 | Conner Chen | -1 word(s) | 2878 | 2022-12-01 01:38:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yi, S.; Wang, L.; Wang, H.; Ho, M.S.; Zhang, S. α-Syn Structure, Aggregation, and Degradation in Parkinson’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/37247 (accessed on 26 July 2026).
Yi S, Wang L, Wang H, Ho MS, Zhang S. α-Syn Structure, Aggregation, and Degradation in Parkinson’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/37247. Accessed July 26, 2026.
Yi, Shuanglong, Linfang Wang, Honglei Wang, Margaret S. Ho, Shiping Zhang. "α-Syn Structure, Aggregation, and Degradation in Parkinson’s Disease" Encyclopedia, https://encyclopedia.pub/entry/37247 (accessed July 26, 2026).
Yi, S., Wang, L., Wang, H., Ho, M.S., & Zhang, S. (2022, November 30). α-Syn Structure, Aggregation, and Degradation in Parkinson’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/37247
Yi, Shuanglong, et al. "α-Syn Structure, Aggregation, and Degradation in Parkinson’s Disease." Encyclopedia. Web. 30 November, 2022.
Copy Citation
Parkinson’s disease (PD) is a progressive neurodegenerative disorder. The classical behavioral defects of PD patients involve motor symptoms such as bradykinesia, tremor, and rigidity, as well as non-motor symptoms such as anosmia, depression, and cognitive impairment. Pathologically, the progressive loss of dopaminergic (DA) neurons in the substantia nigra (SN) and the accumulation of α-synuclein (α-syn)-composed Lewy bodies (LBs) and Lewy neurites (LNs) are key hallmarks.
Parkinson’s disease
α-synuclein
glia
neuron-glia crosstalk
1. Introduction
1.1. The History and Pathogenesis of Parkinson’s Disease
Parkinson’s disease (PD) is named after the English science medical expert James Parkinson, who wrote down the first detailed description about PD in An Essay on the Shaking Palsy in 1817 [1]. With life expectancy increasing, the incidence of PD is considerably rising. There are about 7 million people in pain from PD in the world, of which the United States accounts for about 1 million [2]. In industrialized countries, the morbidity of this disease is about 0.3% and patients are mainly elder adults. For people older than 60, the morbidity sharply increases 1% every year, for people older than 80, the increase rate reached 4% [3].
Upon ageing, PD patients exhibit worsening central nervous system (CNS) symptoms cultivating to defects in the motor system. The pathological hallmark of PD is the progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) [4] (Figure 1D). These DA neurons are required for normal motor function, the death of which lead to bradykinesia, tremor, and rigidity [5]. Another pathological feature of PD is the formation of Lewy bodies (LBs) and Lewy neurites (LNs), which are cytoplasmic spherical protein inclusion bodies mainly composed of α-synuclein (α-syn) [6] (Figure 1D). Importantly, the spread pattern of LBs pathology correlates with the progression of PD clinical symptoms, which is also the basis of the Braak staging system [7][8]. The formation of LBs always is connected with the induction of reactive oxygen species (ROS) and the generation of superoxide radical anions, hydrogen peroxide, and hydroxyl radicals [9]. The accumulating evidence suggests that the increase in oxidative stress would exacerbate the development of PD [10].
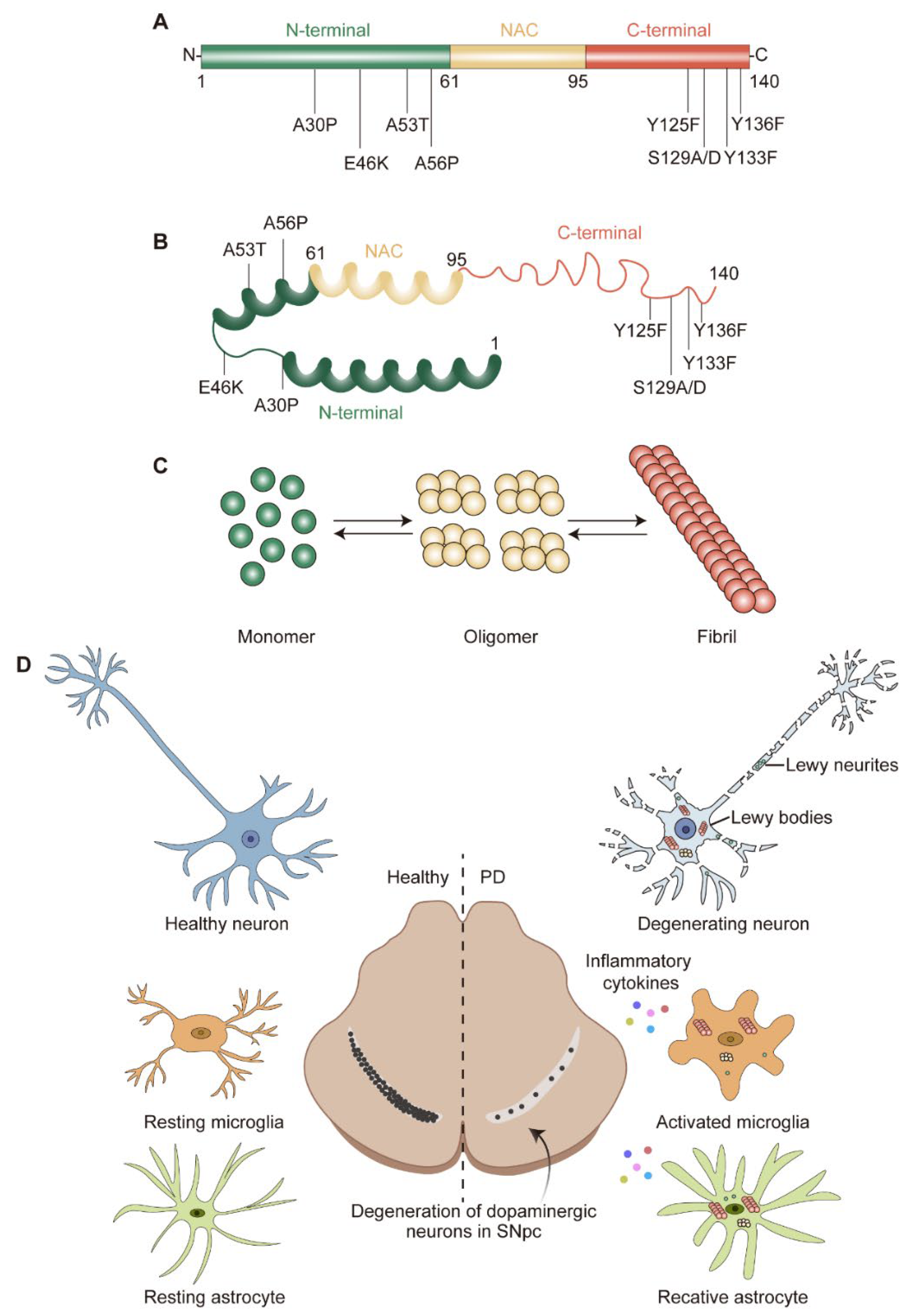
Figure 1. α-syn structure and pathological hallmarks of PD: (A) Schematic representation of α-syn. α-syn is divided into N-terminal, non-amyloid-beta component (NAC), and C-terminal; three domains highlighted in green, yellow, and red, respectively. Several familial PD-related mutations and post-translational modification sites are denoted. (B) Structure of α-syn monomer. (C) α-syn equilibrium. α-syn monomer can aggregate into oligomer or fibril. (D) Pathological hallmarks of PD. The pathological hallmarks of PD include progress loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc), misfolded α-syn aggregates and neurites known as Lewy bodies (LBs) and Lewy neurites (LNs), and glial activation. α-syn could transfer to and activate microglia and astrocyte, which in turn release pro-/anti-inflammatory cytokines and contribute to neurodegeneration.
Although the primary cause of PD cases appears to be spontaneous and widespread, most experts agree that the pathophysiology of this disease is profoundly influenced by the combination of genetic and environmental factors and that how genes and the environment interact can be very complicated. The evidence consistently suggests that a higher risk of PD is associated with a number of environmental factors, including the area of residence, occupation, exposure to metals, pesticide and herbicide exposure, and so on [11]. Accordingly, genes including SCNA [12], Parkin [13], leucine-rich repeat kinase 2 (LRRK2) [14], phosphatase and tensin homolog deleted on chromosome 10-induced putative kinase 1 (PINK1) [15], glucocerebrosidase (GBA) [16], vacuolar protein sorting 35 (VPS35) [17], and DJ-1 [18] are linked to genetic variants that directly contribute to PD. Genome-wide association studies (GWAS) also suggest that both adaptive and innate immunity may play a role in PD pathogenesis [19]. These genes help us to comprehend PD processes at cellular and molecular levels.
1.2. Role of Glia in PD
Although glia outnumber neurons in the CNS, they were originally considered to be the inert “glue” (Greek “glia”) that fill in the space between neurons and play a passive supporting role due to the lack of electrical excitability [20][21][22]. Nonetheless, in the past decades, numerous studies demonstrated that glia maximize their contact with neurons and actively contribute to almost every aspect of neuronal development and function, including neurogenesis, axon guidance and ensheathment, synaptic connection and plasticity, trophic supports, elimination of dying neurons, maintaining ionic balance, and blood–brain barrier (BBB) formation [21][23][24][25][26][27][28][29][30][31]. Importantly, glial cells have been implicated in a series of neurodegenerative diseases including PD [32][33] and many PD risk genes are also expressed in glial cells [34][35][36][37], further shedding light on the importance of glia in maintaining neuronal homeostasis.
2. α-Syn Structure, Aggregation, and Degradation
2.1. α-Syn Structure and Physiological Function
The genetic era in PD research began in 1997, when α-syn was recognized as a key factor in this complicated neurological disease [12]. The protein α-syn is encoded by SNCA and consists of 140 amino acids (a.a.) with a molecular weight of approximately 15 kDa [38]. According to the physiochemical property, α-syn could be divided into three domains: a positively charged N-terminal region (1–60 a.a.) containing four regions of 11 imperfect repeats with the KTKGEV consensus sequence [39]; a central hydrophobic region (61–95 a.a.) with the non-amyloid-beta component (NAC) [40]; and a C-terminal region (96–140 a.a.) enriched with acid residues [41] (Figure 1A). α-syn is widely expressed at the presynaptic terminals of the brain [42] and regulates the vesicular transport of neurotransmitters. Under physiological conditions, α-syn is mostly found in the substantia nigra (SN), cortex, and hippocampus; it is crucial for regulating the function and plasticity of synapses [43]. Despite being highly enriched in the nervous system [42], α-syn is also expressed in a variety of other tissues, including red blood cells, heart, muscle, and other tissues with low expression levels [44], suggesting that α-syn has cellular functions beyond those specific to the nervous system.
At the cellular level, α-syn is localized to the synaptic terminal [42], mitochondria [45], endoplasmic reticulum (ER) [46], Golgi apparatus (GA) [47], endo-lysosome system [48], and also the neuronal nuclei [49]. The name synuclein combines its location in synaptic vesicles (“syn”) and nuclear envelope (“nuclein”). Up to now, there is a limited understanding about the function and physiological role of α-syn in each subcellular compartment. α-syn is linked to presynaptic terminals, sustains normal SNARE-complex assembly, controls the dopamine release [50][51], and promotes membrane curvature during synaptic vesicle budding and trafficking [42]. Increased levels of toxic α-syn cause increased mitochondrial fragmentation and decreased protein import [52][53]. When overexpressing wild-type or mutant α-syn, ER stress increases and calcium homeostasis are impaired [54]. α-syn reduces ionic transport and decreases the membrane traffic of GA [55] and deficiencies in axonal transport were linked to GA fragmentation [56]. Although the synaptic function of α-syn is well recognized [49], its role within the nucleus is less understood.
2.2. α-Syn Misfolding and Aggregation
Previously, PD was considered as an aging disease with an unknown specific cause or hereditary component. However, this idea was disproved in the late 1990s when SNCA gene variants were linked to familial, early onset forms of PD [12]. Further study provided that early onset PD are caused by duplication, triplication, and autosomal dominant missense mutations in the SNCA gene [57][58]. Now, it has been agreed that α-syn misfolding and subsequent aggregation contribute significantly to DA neuron degeneration in PD. This is complicated by a fast-aging global population, which coincides with an increase in the number of sporadic occurrences of PD [59][60].
Although it has long been assumed that α-syn has a natively unfolded tertiary structure [61], monomer α-syn appears to be the predominant species in the brain [62]. Alternatively, α-syn may exist as an α-helically folded tetramer [63]. Both the monomer and tetramer species are resistant to fibrillization and are present in equilibrium within healthy neurons [61][63]. Misfolded α-syn monomers could form oligomers and fibrils, which then aggregate as LBs [64] (Figure 1B,C). Particularly, the misfolding and aggregation of α-syn into fibrils depend on 71–82 a.a. in the central hydrophobic region [65]. This region can aggregate on its own and deletion of 71–82 a.a. or 66–74 a.a. prevents protein aggregation [65][66], indicating that these residues are crucial for protein misfolding and may even be the cause of amyloidosis. This feature was only observed in α-syn and as the concentration of α-syn increases, the propensity to aggregate increases.
Gene mutations in SNCA are also thought to promote α-syn aggregation [67]. The first specific mutation is the A53T substitution, which was an autosomal-dominant single base pair change [12]. Since then, more and more familial PD-causing autosomal dominant SNCA gene mutations have been identified, including A30P [68], E46K [69], H50Q [70], G51D [71], A53E [72], A53V [73], A56P [74], Y133F [75], and Y136F [75] (Figure 1A,B). While the A30P, G51D, and A53E mutations appear to slow down the rate of fibril formation, the E46K, H50Q, and A53T mutations lead to an increased rate of fibril formation. Importantly, studies on these mutants provide compelling evidence that α-syn oligomers and pre-fibrils are more toxic than mature aggregated fibrils and the aggregation of α-syn occurs in early onset PD.
The solubility and aggregation property of α-syn was also affected by post-translational modifications (PTMs), such as phosphorylation, ubiquitination, nitration, truncation, and O-GlcNAcylation [76][77]. Among these PTMs, phosphorylation at residue S129 (α-synpS129) and its potential connection to α-syn-induced neurodegeneration have drawn intensive attention [41][78]. LBs in PD patient brains exists in dramatically higher amounts of α-synpS129 than for normal conditions [79]. In a Drosophila PD model, α-synpS129 was observed when expressing wild-type or mutant α-syn and the phosphorylation preferences are A53T > A30P > wild-type α-syn [80]. Further studies showed that the phosphorylation-resistant S129A mutant reduces the toxicity caused by α-syn, while the S129D mutant increases it [81]. Interestingly, other researchers also found that α-synpS129 showed a reduced aggregation propensity and cytotoxicity in yeast and in vitro [82][83]. This may be explained by findings that long-range interactions could stabilize the conformation of monomeric α-syn and act as an inhibitor of oligomerization and aggregation [84], while α-synpS129 could disrupt this interaction [85]. According to a recent report, α-syn at Tyr125 also can be phosphorylated (α-synpTyr125) and this phosphorylation occurs at a young age but declines during the aging process. Preventing Tyr125 phosphorylation might cause α-syn to be more toxic [86]. These authors demonstrate that phosphorylation at Ser129 increases, while phosphorylation at Tyr125 decreases, the soluble oligomer of α-syn [86]. Thus, in most cases, Ser129 phosphorylation positively regulates α-syn toxicity by accelerating oligomer formation; conversely, Tyr125 phosphorylation negatively regulates α-syn toxicity by inhibiting oligomer formation.
2.3. α-Syn Degradation
The pathogenesis of PD and associated synucleinopathies depend heavily on the levels and conformation of α-syn. α-syn has been found to be regulated by homeostatic mechanisms via protein secretion and degradation at various points in both intracellular and transcellular ways [87]. Therefore, understanding the removal of various forms of α-syn is essential for PD pathogenesis and potential treatments.
Intracellular proteins are primarily degraded by proteasomal and lysosomal pathways. Proteasome degrades intracellular proteins through the ubiquitin-proteasome system (UPS), which involves the chain-like conjugation of at least four ubiquitin molecules on lysine residues of substrate proteins [88]. Lysosome degrades intracellular proteins via autophagy-lysosome pathways (ALP), including macro-autophagy (also known as autophagy), chaperone-mediated autophagy (CMA), and micro-autophagy [89]. Despite intensive studies, the exact mechanism for α-syn degradation remains controversial and varies depending on the system used. Both proteasome and lysosome were shown to be able to degrade recombinant α-syn in in vitro purified systems [90]. Accordingly, the monomeric, dimeric, pre-fibrillar, and mutant α-syn are preferentially degraded through UPS; whereas CMA degrades monomeric or dimeric α-syn, and the only way to degrade oligomeric and aggregated α-syn is autophagy. Under normal conditions, UPS is the main degradation pathway, but under stress or pathological conditions, autophagy and CMA pathways are recruited to clear the toxic α-syn burden (Figure 2).
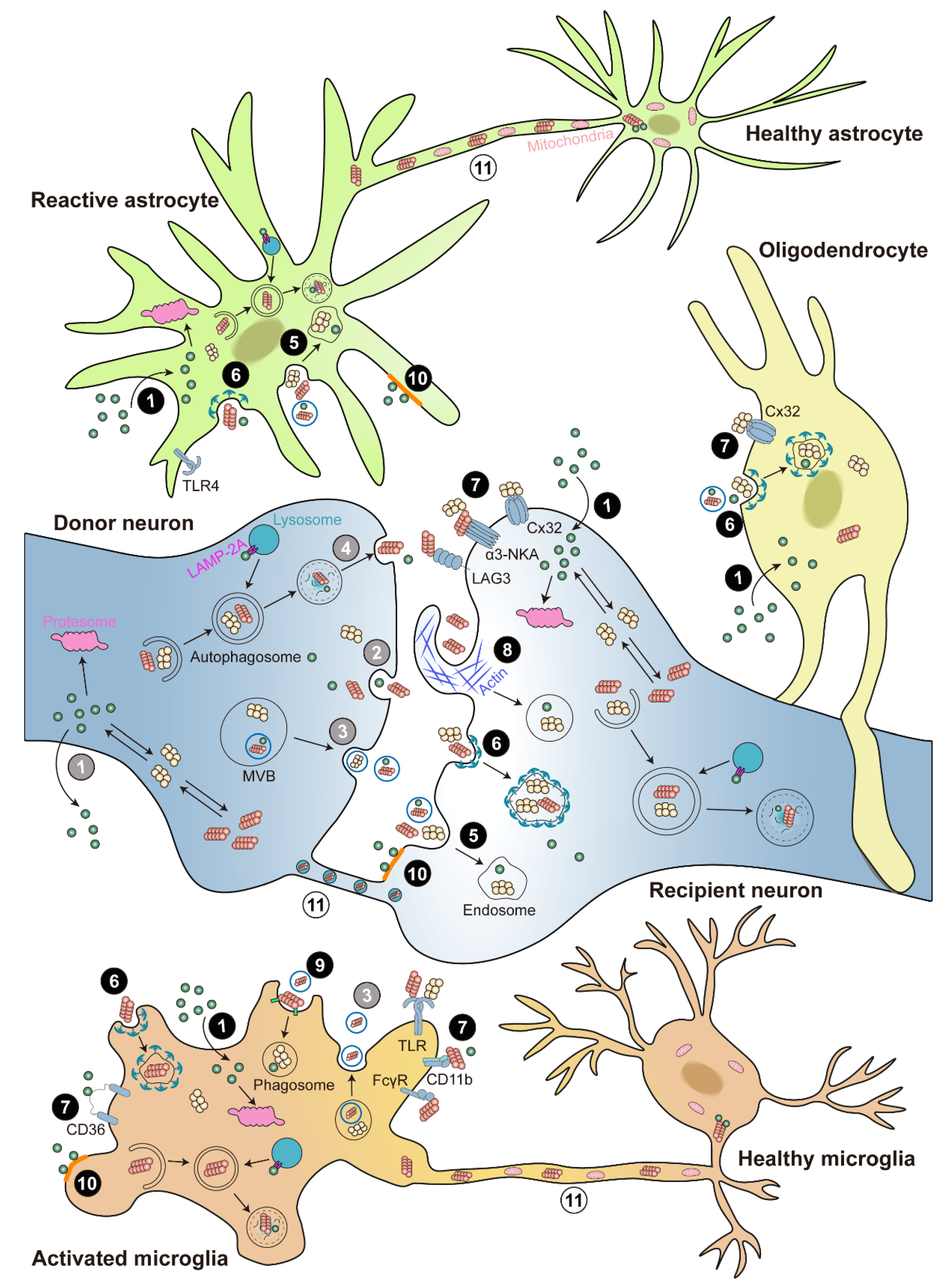
Figure 2. Cell-to-cell transmission of α-syn. Illustration of α-syn cell-to-cell transmission. α-syn could transfer between neurons and glia. α-syn is released via ① passive diffusion (only monomer), ② exocytosis, ③ exosomes, or ④ exophagy (grey color numbers). α-syn is taken up via ① passive diffusion (only monomer), ⑤ endocytosis, ⑥ clathrin-mediated endocytosis (CME), ⑦ receptor-mediated internalization, ⑧ micropinocytosis, ⑨ phagocytosis, or ⑩ lipid raft (black color numbers). The receptors involved in α-syn internalization include lymphocyte-activation gene 3 (LAG3), α3-subunit of Na+/K+-ATPase (α3-NKA), and the gap junction protein connexin-32 (Cx32) in neuron; Toll-like receptors 2 and 4 (TLR2 and TLR4), the scavenger receptor CD36, integrin CD11b, and the Fcγ receptors (FcγR) in microglia; and Cx32 in oligodendrocyte. In addition, α-syn can directly cell-to-cell transfer by ⑪ tunneling nanotubes (TNTs) (white color numbers). Internalized α-syn are degraded via the ubiquitin-proteasome system (UPS), autophagy, and chaperone-mediated autophagy (CMA) pathways.
2.3.1. α-Syn Degradation through UPS
The increased accumulation of non-ubiquitinated and ubiquitinated proteins in the LBs of PD patients, as well as the reduced expression of 20S and 26S UPS subunits, suggested that proteasome was involved in the toxic manifestations of PD [88][91]. Both in vitro and in vivo studies show that α-syn is degraded by proteasomes, with not only monomeric, but also possibly pre-fibrillar α-syn species [92][93]. Due to differential effects on cellular α-syn half-life, a number of Ubi-ligases catalyze the addition of mono- or poly-Ubi chains to α-syn, with cytoprotective or toxic effects depending on the specific experimental setup [92][94][95].
The studies about the degradation of phosphorylated α-syn are contradictory. The research in cultured cells and rat primary cortical cultures revealed that the proteasome system degrades α-synpS129 in a Ubi-independent pathway [96][97][98]. However, the lysosome inhibitor could lead to accumulated α-synpS129 by blocking the ALP [99][100]. Using a synthesis inhibitor (cycloheximide), they found that the half-life time of α-synpS129 is significantly shorter than the non-phosphorylated form, implying that the phosphorylated form is selectively targeted for degradation, and inhibition of proteasome significantly prolonged the half-life time of α-synpS129 [98]. These findings suggest that monomeric α-synpS129 may be degraded primarily by the proteasome and that lysosome plays a compensatory role.
Notably, multiple lines of evidence point to a possibility that elevated α-syn inhibits proteasomal activity, which could then cause an increase in α-syn levels, thus creating a pathogenic feedback loop that favors α-syn aggregation [101][102]. Understood together, only specific types of α-syn, consisting of tiny, soluble oligomers, are degraded by the UPS pathway.
2.3.2. α-Syn Degradation through ALP
The role of proteasome in α-syn degradation are in debate. Investigations in numerous cellular systems found no significant accumulation of endogenous or overexpressed α-syn with proteasomal inhibition, while the inhibition or genetic depletion of proteasome in a mice model show increased and accumulated α-syn [103][104]. One possible explanation of the dispute is the assembly state and pools of α-syn exploited, since large oligomeric forms can be removed exclusively by lysosomes but not UPS [48]. According to those reports, a large fraction of α-syn were degraded via the lysosomal pathways [105][106] (Figure 2). The major evidence for autophagy in α-syn degradation comes from studies that α-syn buildup was detected after exposing cell lines to 3-methyladenine, an autophagosome formation inhibitor, when overexpressing either wild-type or mutant α-syn [90][107]. However, it is currently controversial whether aggregated, insoluble α-syn assemblies, such as those found in LBs, can be broken down by autophagy [108]. For instance, in an exogenous α-syn pre-formed fibrils (PFF) triggered endogenous an α-syn aggregation cell model, the α-syn inclusion resisted lysosomal breakdown. However, Gao and colleagues recently showed that distinct autophagy inducers resulted in the increased destruction of ingested exogenous α-syn PFF in neuronal cell lines, indicating that lysosomes may be able to remove seeded fibrillar α-syn [109]. Besides, it has been demonstrated that increasing autophagy flux upon α-syn overexpression has negative effects [107][110], including enhanced secretion of α-syn assemblies into the extracellular space, which may aid in the spreading of pathogenic α-syn, and an increase in mitochondrial degradation (mitophagy) in both cellular and animal PD models [111].
The above contradictions suggested that an alternative lysosomal mechanism might exist in α-syn degradation, as nonspecific lysosomal inhibitors had more dramatic effects than selective autophagy inhibition in some cases. This particular problem had actually been answered since the late 1980s, almost entirely through the work of Fred Dice’s lab, which confirmed the existence of CMA by a series of C. elegans experiments [112]. The proteins intended for CMA typically contain the targeting motif KFERQ and are selectively translocated into the autophagic pathway rather than being sequestered through bulk engulfment of cytoplasmic contents [113]. They are identified by the cytosolic chaperone heat-shock cognate 70 (Hsc70) and then transported directly into lysosomes by interaction with the lysosomes-associated membrane protein 2A (LAMP-2A) [114][115]. The involvement of CMA in α-syn degradation is supported by a number lines of evidence. The C-terminal KFERQ-like motif in α-syn could bind to LAMP-2A for CMA degradation, while for A30P and A53T mutant α-syn, although binding to LAMP-2A was not affected, they were not able to be internalized or degraded, acting as inhibitors of CMA degradation of other substrates [106][116][117]. This significant discovery has sparked curiosity in the relationship between CMA and PD pathogenesis. Lower levels of LAMP-2A were found in early-stage PD patient brains compared to healthy controls, along with an accumulation of α-syn and other CMA substrates such as myocyte-specific enhancer factor 2D (MEF2D) [106][118]. Consistently, downregulating LAMP-2A expression in rats results in ubiquitin-positive α-syn inclusions accumulated in SN, followed by DA neuronal death [119]. Some forms of PTMs, such as oxidation, nitration, and modification by oxidized dopamine, could hinder α-syn degradation via CMA and cause its accumulation [120]. Dopamine-modified α-syn (seen in sporadic PD patients) also obstructs the processing of other CMA substrates, very similar to A30P and A53T mutant α-syn [116][120]. Interestingly, other studies show that the complete loss of LAMP-2A in the murine brain did not induce significant α-syn accumulation, neither monomer nor high-molecular weight species [121]; this may result from the above-mentioned other protein degradation processes such as UPS.
References
- Parkinson, J. An essay on the shaking palsy, 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236.
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. npj Park. Dis. 2018, 4, 21.
- de Rijk, M.C.; Breteler, M.M.; Graveland, G.A.; Ott, A.; Grobbee, D.E.; van der Meche, F.G.; Hofman, A. Prevalence of Parkinson’s disease in the elderly: The Rotterdam Study. Neurology 1995, 45, 2143–2146.
- Barbeau, A. L-dopa therapy in Parkinson’s disease: A critical review of nine years’ experience. Can. Med. Assoc. J. 1969, 101, 59–68.
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601.
- McKeith, I.G.; Burn, D.J.; Ballard, C.G.; Collerton, D.; Jaros, E.; Morris, C.M.; McLaren, A.; Perry, E.K.; Perry, R.; Piggott, M.A. Dementia with Lewy bodies. Semin. Clin. Neuropsychiatry 2003, 8, 46–57.
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211.
- Braak, H.; Muller, C.M.; Rub, U.; Ackermann, H.; Bratzke, H.; de Vos, R.A.; Del Tredici, K. Pathology associated with sporadic Parkinson’s disease—Where does it end? In Parkinson’s Disease and Related Disorders; Riederer, P., Reichmann, H., Youdim, M.B.H., Gerlach, M., Eds.; Journal of Neural Transmission. Supplementa Series; Springer: Vienna, Austria, 2006; pp. 89–97.
- Cookson, M.R. The biochemistry of Parkinson’s disease. Annu. Rev. Biochem. 2005, 74, 29–52.
- Filomeni, G.; Graziani, I.; De Zio, D.; Dini, L.; Centonze, D.; Rotilio, G.; Ciriolo, M.R. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: Possible implications for Parkinson’s disease. Neurobiol. Aging 2012, 33, 767–785.
- Chen, H.; Ritz, B. The search for environmental causes of Parkinson’s disease: Moving forward. J. Park. Dis. 2018, 8, S9–S17.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Haywood, A.F.; Staveley, B.E. Parkin counteracts symptoms in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2004, 5, 14.
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797.
- Clark, I.E.; Dodson, M.W.; Jiang, C.G.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166.
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.Y.; Whitworth, A.J.; Schapira, A.H. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380.
- Vilarino-Guell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167.
- Meulener, M.; Whitworth, A.J.; Armstrong-Gold, C.E.; Rizzu, P.; Heutink, P.; Wes, P.D.; Pallanck, L.J.; Bonini, N.M. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr. Biol. 2005, 15, 1572–1577.
- Pierce, S.; Coetzee, G.A. Parkinson’s disease-associated genetic variation is linked to quantitative expression of inflammatory genes. PLoS ONE 2017, 12, e0175882.
- Kremer, M.C.; Jung, C.; Batelli, S.; Rubin, G.M.; Gaul, U. The glia of the adult, D.rosophila nervous system. Glia 2017, 65, 606–638.
- Losada-Perez, M. Glia: From ‘just glue’ to essential players in complex nervous systems: A comparative view from flies to mammals. J. Neurogenet. 2018, 32, 78–91.
- Fields, R.D.; Stevens-Graham, B. New insights into neuron-glia communication. Science 2002, 298, 556–562.
- Wilton, D.K.; Dissing-Olesen, L.; Stevens, B. Neuron-Glia Signaling in Synapse Elimination. Annu. Rev. Neurosci. 2019, 42, 107–127.
- Ou, J.; He, Y.; Xiao, X.; Yu, T.M.; Chen, C.; Gao, Z.; Ho, M.S. Glial cells in neuronal development: Recent advances and insights from Drosophila melanogaster. Neurosci. Bull. 2014, 30, 584–594.
- Yang, M.; Wang, H.; Chen, C.; Zhang, S.; Wang, M.; Senapati, B.; Li, S.; Yi, S.; Wang, L.; Zhang, M.; et al. Glia-derived temporal signals orchestrate neurogenesis in the Drosophila mushroom body. Proc. Natl. Acad. Sci. USA 2021, 118, e2020098118.
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440.
- Chotard, C.; Salecker, I. Neurons and glia: Team players in axon guidance. Trends Neurosci. 2004, 27, 655–661.
- Bainton, R.J.; Tsai, L.T.; Schwabe, T.; DeSalvo, M.; Gaul, U.; Heberlein, U. Moody encodes two GPCRs that regulate cocaine behaviors and blood-brain barrier permeability in Drosophila. Cell 2005, 123, 145–156.
- Blauth, K.; Banerjee, S.; Bhat, M.A. Axonal ensheathment and intercellular barrier formation in Drosophila. Int. Rev. Cell Mol. Biol. 2010, 283, 93–128.
- Eroglu, C.; Barres, B.A. Regulation of synaptic connectivity by glia. Nature 2010, 468, 223–231.
- Stork, T.; Bernardos, R.; Freeman, M.R. Analysis of glial cell development and function in Drosophila. Cold Spring Harb. Protoc. 2012, 2012, pdb-top067587.
- Zuchero, J.B.; Barres, B.A. Glia in mammalian development and disease. Development 2015, 142, 3805–3809.
- Wang, L.; Wang, H.; Yi, S.; Zhang, S.; Ho, M.S. A LRRK2/dLRRK-mediated lysosomal pathway that contributes to glial cell death and DA neuron survival. Traffic 2022, 23, 506–520.
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 2004, 127, 420–430.
- Mullett, S.J.; Hamilton, R.L.; Hinkle, D.A. DJ-1 immunoreactivity in human brain astrocytes is dependent on infarct presence and infarct age. Neuropathology 2009, 29, 125–131.
- Miklossy, J.; Arai, T.; Guo, J.P.; Klegeris, A.; Yu, S.; McGeer, E.G.; McGeer, P.L. LRRK2 expression in normal and pathologic human brain and in human cell lines. J. Neuropathol. Exp. Neurol. 2006, 65, 953–963.
- Kam, T.I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol. Dis. 2020, 144, 105028.
- Venda, L.L.; Cragg, S.J.; Buchman, V.L.; Wade-Martins, R. Alpha-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 2010, 33, 559–568.
- Jao, C.C.; Hegde, B.G.; Chen, J.; Haworth, I.S.; Langen, R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671.
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005, 280, 9595–9603.
- Oueslati, A.; Fournier, M.; Lashuel, H.A. Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: Implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 2010, 183, 115–145.
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32.
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48.
- Nakai, M.; Fujita, M.; Waragai, M.; Sugama, S.; Wei, J.; Akatsu, H.; Ohtaka-Maruyama, C.; Okado, H.; Hashimoto, M. Expression of α-synuclein, a presynaptic protein implicated in Parkinson’s disease, in erythropoietic lineage. Biochem. Biophys. Res. Commun. 2007, 358, 104–110.
- Li, W.-W.; Yang, R.; Guo, J.-C.; Ren, H.-M.; Zha, X.-L.; Cheng, J.-S.; Cai, D.-F. Localization of α-synuclein to mitochondria within midbrain of mice. NeuroReport 2007, 18, 1543–1546.
- Hoozemans, J.J.M.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.I.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711.
- Gosavi, N.; Lee, H.J.; Lee, J.S.; Patel, S.; Lee, S.J. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 2002, 277, 48984–48992.
- Lee, H.J.; Khoshaghideh, F.; Patel, S.; Lee, S.J. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 2004, 24, 1888–1896.
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815.
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667.
- Yavich, L.; Tanila, H.; Vepsalainen, S.; Jakala, P. Role of alpha-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004, 24, 11165–11170.
- Chen, H.; Chan, D.C. Physiological functions of mitochondrial fusion. Ann. N. Y. Acad. Sci. 2010, 1201, 21–25.
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343.
- Belal, C.; Ameli, N.J.; El Kommos, A.; Bezalel, S.; Al’Khafaji, A.M.; Mughal, M.R.; Mattson, M.P.; Kyriazis, G.A.; Tyrberg, B.; Chan, S.L. The homocysteine-inducible endoplasmic reticulum (ER) stress protein Herp counteracts mutant alpha-synuclein-induced ER stress via the homeostatic regulation of ER-resident calcium release channel proteins. Hum. Mol. Genet. 2012, 21, 963–977.
- Fan, J.; Hu, Z.; Zeng, L.; Lu, W.; Tang, X.; Zhang, J.; Li, T. Golgi apparatus and neurodegenerative diseases. Int. J. Dev. Neurosci. 2008, 26, 523–534.
- Fujita, Y.; Ohama, E.; Takatama, M.; Al-Sarraj, S.; Okamoto, K. Fragmentation of Golgi apparatus of nigral neurons with α-synuclein-positive inclusions in patients with Parkinson’s disease. Acta Neuropathol. 2006, 112, 261–265.
- Parsian, A.; Racette, B.; Zhang, Z.H.; Chakraverty, S.; Rundle, M.; Goate, A.; Perlmutter, J.S. Mutation, sequence analysis, and association studies of alpha-synuclein in Parkinson’s disease. Neurology 1998, 51, 1757–1759.
- Polymeropoulos, M.H. Autosomal dominant Parkinson’s disease and alpha-Synuclein. Ann. Neurol. 1998, 44, S63–S64.
- Morgan, J. A seat at the table for people with Parkinson’s disease. Lancet Neurol. 2015, 14, 1077–1078.
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953.
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802.
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. Alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364.
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110.
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 2003, 37, 583–595.
- Waxman, E.A.; Mazzulli, J.R.; Giasson, B.I. Characterization of hydrophobic residue requirements for α-synuclein fibrillization. Biochemistry 2009, 48, 9427–9436.
- Du, H.N.; Tang, L.; Luo, X.Y.; Li, H.T.; Hu, J.; Zhou, J.W.; Hu, H.Y. A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human alpha-synuclein. Biochemistry 2003, 42, 8870–8878.
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846.
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J. Mol. Biol. 2002, 315, 799–807.
- Choi, W.; Zibaee, S.; Jakes, R.; Serpell, L.C.; Davletov, B.; Crowther, R.A.; Goedert, M. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 2004, 576, 363–368.
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q.; a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813.
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471.
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5.
- Yoshino, H.; Hirano, M.; Stoessl, A.J.; Imamichi, Y.; Ikeda, A.; Li, Y.; Funayama, M.; Yamada, I.; Nakamura, Y.; Sossi, V.; et al. Homozygous alpha-synuclein p.A53V in familial Parkinson’s disease. Neurobiol. Aging 2017, 57, 248.e7–248.e12.
- Karpinar, D.P.; Balija, M.B.G.; Kügler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H. Pre-fibrillar α-synuclein variants with impaired β-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 2009, 28, 3256–3268.
- Negro, A.; Brunati, A.M.; Donella-Deana, A.; Massimino, M.L.; Pinna, L.A. Multiple phosphorylation of alpha-synuclein by protein tyrosine kinase Syk prevents eosin-induced aggregation. FASEB J. 2002, 16, 210–212.
- Mavroeidi, P.; Xilouri, M. Neurons and Glia Interplay in alpha-Synucleinopathies. Int. J. Mol. Sci. 2021, 22, 4994.
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091.
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein phosphorylation in neurodegeneration: Friend or foe? Front. Mol. Neurosci. 2014, 7, 42.
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164.
- Takahashi, M.; Kanuka, H.; Fujiwara, H.; Koyama, A.; Hasegawa, M.; Miura, M.; Iwatsubo, T. Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci. Lett. 2003, 336, 155–158.
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663.
- Tenreiro, S.; Reimao-Pinto, M.M.; Antas, P.; Rino, J.; Wawrzycka, D.; Macedo, D.; Rosado-Ramos, R.; Amen, T.; Waiss, M.; Magalhaes, F.; et al. Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLoS Genet. 2014, 10, e1004302.
- Ghanem, S.S.; Majbour, N.K.; Vaikath, N.N.; Ardah, M.T.; Erskine, D.; Jensen, N.M.; Fayyad, M.; Sudhakaran, I.P.; Vasili, E.; Melachroinou, K.; et al. Alpha-Synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc. Natl. Acad. Sci. USA 2022, 119, e2109617119.
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435.
- Paleologou, K.E.; Schmid, A.W.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fredenburg, R.; Lansbury, P.T., Jr.; Fernandez, C.; Eliezer, D.; Zweckstetter, M.; et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J. Biol. Chem. 2008, 283, 16895–16905.
- Chen, L.; Periquet, M.; Wang, X.; Negro, A.; McLean, P.J.; Hyman, B.T.; Feany, M.B. Tyrosine and serine phosphorylation of α-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Investig. 2009, 119, 3257–3265.
- Tofaris, G.K.; Goedert, M.; Spillantini, M.G. The Transcellular Propagation and Intracellular Trafficking of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2017, 7, a024380.
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46.
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849.
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013.
- McNaught, K.S.P.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194.
- Shimura, H.; Schlossmacher, M.C.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269.
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851.
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328.
- Lee, J.T.; Wheeler, T.C.; Li, L.; Chin, L.S. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 2008, 17, 906–917.
- Waxman, E.A.; Giasson, B.I. Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J. Neuropathol. Exp. Neurol. 2008, 67, 402–416.
- Tofaris, G.K.; Layfield, R.; Spillantini, M.G. Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001, 509, 22–26.
- Machiya, Y.; Hara, S.; Arawaka, S.; Fukushima, S.; Sato, H.; Sakamoto, M.; Koyama, S.; Kato, T. Phosphorylated α-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J. Biol. Chem. 2010, 285, 40732–40744.
- Arawaka, S.; Sato, H.; Sasaki, A.; Koyama, S.; Kato, T. Mechanisms underlying extensive Ser129-phosphorylation in alpha-synuclein aggregates. Acta Neuropathol. Commun. 2017, 5, 48.
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018, 557, 558–563.
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968.
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097.
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J. Neurosci. 2011, 31, 14508–14520.
- Bedford, L.; Hay, D.; Devoy, A.; Paine, S.; Powe, D.G.; Seth, R.; Gray, T.; Topham, I.; Fone, K.; Rezvani, N.; et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 2008, 28, 8189–8198.
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in, Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472.
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556.
- Yu, W.H.; Dorado, B.; Figueroa, H.Y.; Wang, L.; Planel, E.; Cookson, M.R.; Clark, L.N.; Duff, K.E. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric α-synuclein. Am. J. Pathol. 2009, 175, 736–747.
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210.
- Gao, J.; Perera, G.; Bhadbhade, M.; Halliday, G.M.; Dzamko, N. Autophagy activation promotes clearance of alpha-synuclein inclusions in fibril-seeded human neural cells. J. Biol. Chem. 2019, 294, 14241–14256.
- Klucken, J.; Poehler, A.-M.; Ebrahimi-Fakhari, D.; Schneider, J.; Nuber, S.; Rockenstein, E.; Schlötzer-Schrehardt, U.; Hyman, B.T.; McLean, P.J.; Masliah, E. Alpha-synuclein aggregation involves a bafilomycin A1-sensitive autophagy pathway. Autophagy 2012, 8, 754–766.
- Chen, L.; Xie, Z.; Turkson, S.; Zhuang, X. A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J. Neurosci. 2015, 35, 890–905.
- Dice, J.F.; Chiang, H.L.; Spencer, E.P.; Backer, J.M. Regulation of catabolism of microinjected ribonuclease A. Identification of residues 7-11 as the essential pentapeptide. J. Biol. Chem. 1986, 261, 6853–6859.
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075.
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786.
- Huang, C.; Cheng, H.; Hao, S.; Zhou, H.; Zhang, X.; Gao, J.; Sun, Q.H.; Hu, H.; Wang, C.C. Heat shock protein 70 inhibits alpha-synuclein fibril formation via interactions with diverse intermediates. J. Mol. Biol. 2006, 364, 323–336.
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295.
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 2016, 31, 178–192.
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647.
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247.
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.-C.; Follenzi, A. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788.
- Rothaug, M.; Stroobants, S.; Schweizer, M.; Peters, J.; Zunke, F.; Allerding, M.; D’Hooge, R.; Saftig, P.; Blanz, J. LAMP-2 deficiency leads to hippocampal dysfunction but normal clearance of neuronal substrates of chaperone-mediated autophagy in a mouse model for Danon disease. Acta Neuropathol. Commun. 2015, 3, 6.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
3 times
(View History)
Update Date:
01 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No