 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohsen Akbarian | -- | 4189 | 2022-11-29 18:14:14 | | | |
| 2 | Amina Yu | Meta information modification | 4189 | 2022-11-30 02:55:53 | | | | |
| 3 | Amina Yu | -137 word(s) | 4052 | 2022-12-02 01:39:55 | | | | |
| 4 | Amina Yu | + 13 word(s) | 4065 | 2022-12-02 01:51:06 | | |
Video Upload Options
At the beginning of the transcription of a protein, thanks to the intracellular chaperone systems and the biophysical laws governing protein folding, correct folding occurs most of the time. When due to cellular defects and rapid protein expression, protein folding becomes problematic, and several fates may occur for the protein. In cases where misfolding leads to the loss of protein activity (such as enzymes), the corresponding disease will appear directly. As this state continues, the misfolded protein may turn into amorphous clots or aggregates with regular structures, each of which can lead to various neurological diseases or even cancer. In the most optimistic scenario, the misfolded protein enters the proteasome machinery and is initially converted into smaller peptides and finally broken down into building amino acids.
1. Genetic Engineering: Protein Analogs
1.1. Site-Directed Mutagenesis
1.2. Fusion Strategies
2. Covalent Engineering
2.1. Protein–Polymer Conjugates
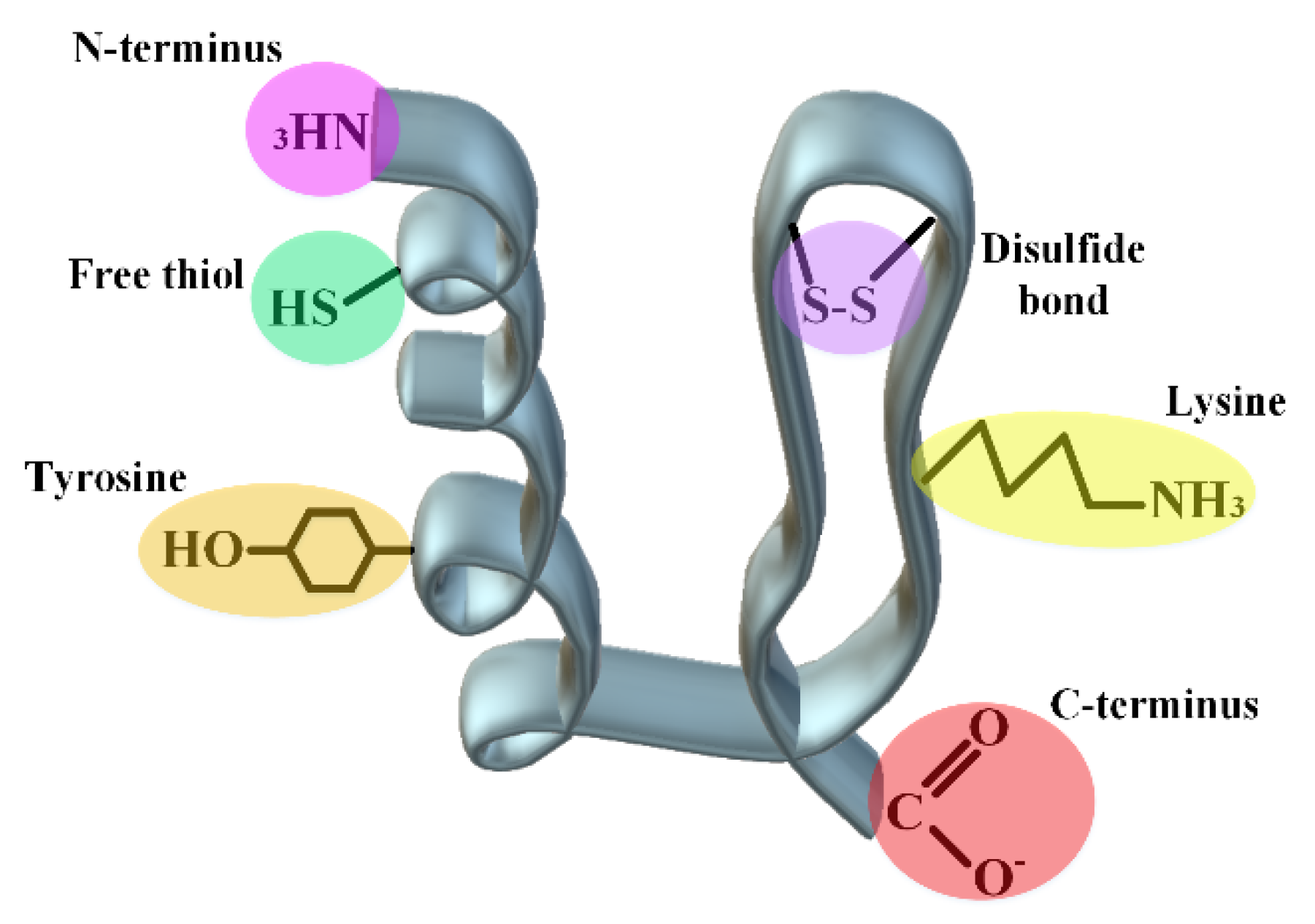
2.2. Linker Chemistry
2.3. Acylation
2.4. Cyclization
2.5. Nanoparticles; Double-Blade Sword
3. Non-Covalent Engineering of Therapeutic Proteins and Peptides; Solvent Engineering Pathways
3.1. Formulation: Solvent Engineering Pathways
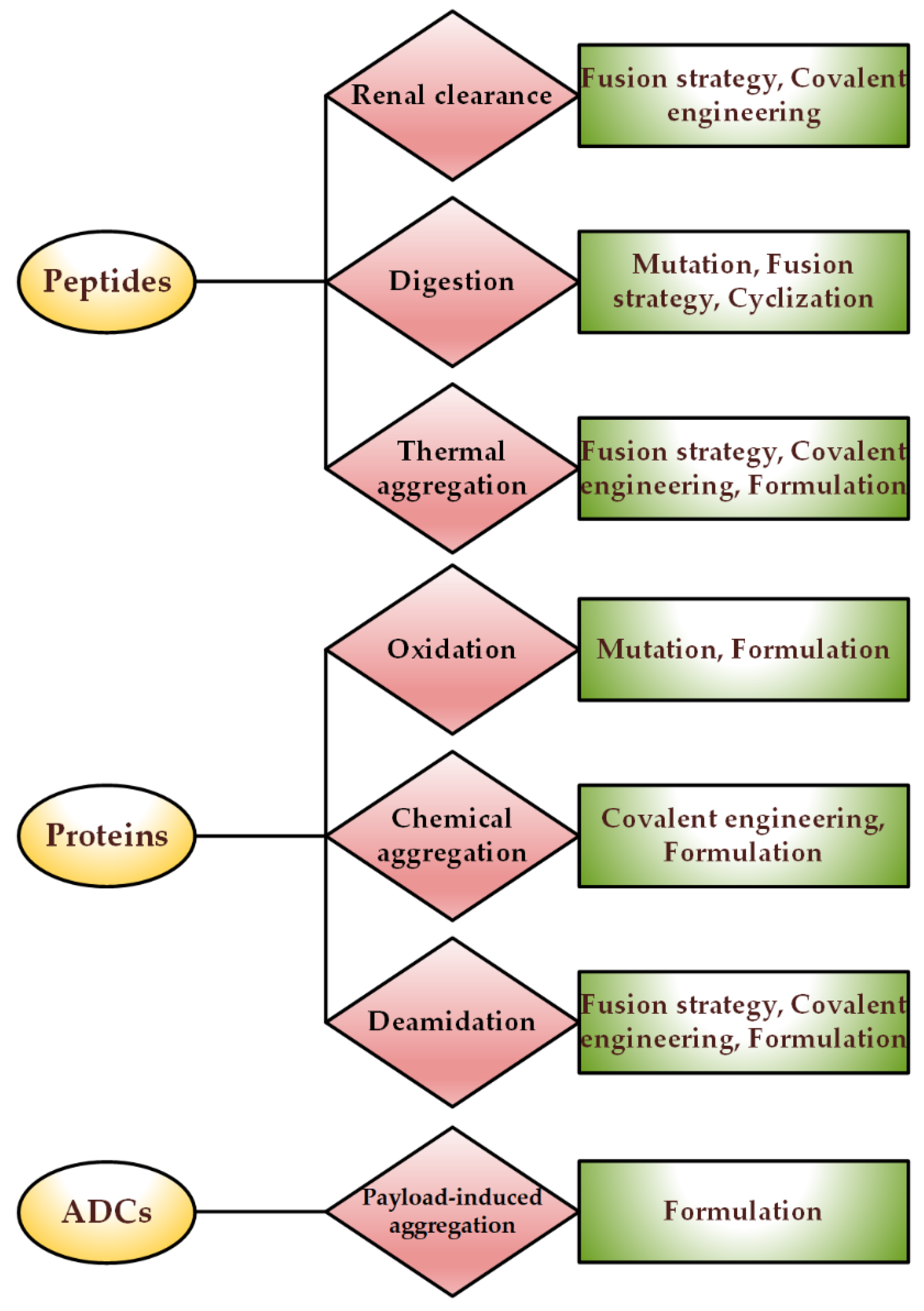
3.2. Choice of Container
References
- Akbarian, M. Insulin therapy: A valuable legacy and its future perspective. Int. J. Biol. Macromol. 2021, 181, 1224–1230.
- Akbarian, M.; Ghasemi, Y.; Uversky, V.N.; Yousefi, R. Chemical modifications of insulin: Finding a compromise between stability and pharmaceutical performance. Int. J. Pharm. 2018, 547, 450–468.
- Akbarian, M.; Yousefi, R.; Moosavi-Movahedi, A.A.; Ahmad, A.; Uversky, V.N. Modulating insulin fibrillation using engineered B-chains with mutated C-termini. Biophys. J. 2019, 117, 1626–1641.
- Arakawa, T.; Prestrelski, S.J.; Narhi, L.O.; Boone, T.C.; Kenney, W.C. Cysteine 17 of recombinant human granulocyte-colony stimulating factor is partially solvent-exposed. J. Protein Chem. 1993, 12, 525–531.
- Culajay, J.F.; Blaber, S.I.; Khurana, A.; Blaber, M. Thermodynamic characterization of mutants of human fibroblast growth factor 1 with an increased physiological half-life. Biochemistry 2000, 39, 7153–7158.
- Luo, P.; Hayes, R.J.; Chan, C.; Stark, D.M.; Hwang, M.Y.; Jacinto, J.M.; Juvvadi, P.; Chung, H.S.; Kundu, A.; Ary, M.L.; et al. Development of a cytokine analog with enhanced stability using computational ultrahigh throughput screening. Protein Sci. 2002, 11, 1218–1226.
- Marshall, S.A.; Lazar, G.A.; Chirino, A.J.; Desjarlais, J.R. Rational design and engineering of therapeutic proteins. Drug Discov. Today 2003, 8, 212–221.
- Pipe, S.W.; Kaufman, R.J. Characterization of a genetically engineered inactivation-resistant coagulation factor VIIIa. Proc. Natl. Acad. Sci. USA 1997, 94, 11851–11856.
- Sytkowski, A.J.; Lunn, E.D.; Risinger, M.A.; Davis, K.L. An erythropoietin fusion protein comprised of identical repeating domains exhibits enhanced biological properties. J. Biol. Chem. 1999, 274, 24773–24778.
- Strohl, W.R. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. Biodrugs 2015, 29, 215–239.
- Marques, J.A.; George, J.K.; Smith, I.J.; Bhakta, V.; Sheffield, W.P. A barbourin-albumin fusion protein that is slowly cleared in vivo retains the ability to inhibit platelet aggregation in vitro. Thromb. Haemost. 2001, 86, 902–908.
- Syed, S.; Schuyler, P.D.; Kulczycky, M.; Sheffield, W.P. Potent antithrombin activity and delayed clearance from the circulation characterize recombinant hirudin genetically fused to albumin. Blood J. Am. Soc. Hematol. 1997, 89, 3243–3252.
- Dennis, M.S.; Zhang, M.; Meng, Y.G.; Kadkhodayan, M.; Kirchhofer, D.; Combs, D.; Damico, L.A. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem. 2002, 277, 35035–35043.
- Akbarian, M.; Yousefi, R. Human αB-crystallin as fusion protein and molecular chaperone increases the expression and folding efficiency of recombinant insulin. PLoS ONE 2018, 13, e0206169.
- Khosravi, F.; Upadhyay, M.; Kumar, A.; Shahsavani, M.B.; Akbarian, M.; Najafi, H.; Tamaddon, A.M.; Yousefi, R. A novel method for the chaperone aided and efficient production of human proinsulin in the prokaryotic system. J. Biotechnol. 2022, 346, 35–46.
- Ko, J.H.; Maynard, H.D. A guide to maximizing the therapeutic potential of protein–polymer conjugates by rational design. Chem. Soc. Rev. 2018, 47, 8998–9014.
- Pelegri-O’Day, E.M.; Lin, E.-W.; Maynard, H.D. Therapeutic protein–polymer conjugates: Advancing beyond PEGylation. J. Am. Chem. Soc. 2014, 136, 14323–14332.
- Rosen, C.B.; Francis, M.B. Targeting the N terminus for site-selective protein modification. Nat. Chem. Biol. 2017, 13, 697–705.
- Kuan, S.L.; Wang, T.; Weil, T. Site-Selective Disulfide Modification of Proteins: Expanding Diversity beyond the Proteome. Chem.–A Eur. J. 2016, 22, 17112–17129.
- Kinstler, O.; Molineux, G.; Treuheit, M.; Ladd, D.; Gegg, C. Mono-N-terminal poly (ethylene glycol)–protein conjugates. Adv. Drug Deliv. Rev. 2002, 54, 477–485.
- Qi, Y.; Amiram, M.; Gao, W.; McCafferty, D.G.; Chilkoti, A. Sortase-catalyzed initiator attachment enables high yield growth of a stealth polymer from the C terminus of a protein. Macromol. Rapid Commun. 2013, 34, 1256–1260.
- Stephanopoulos, N.; Francis, M.B. Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol. 2011, 7, 876–884.
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody–drug conjugates: Recent advances in linker chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907.
- Sano, K.; Nakajima, T.; Miyazaki, K.; Ohuchi, Y.; Ikegami, T.; Choyke, P.L.; Kobayashi, H. Short PEG-linkers improve the performance of targeted, activatable monoclonal antibody-indocyanine green optical imaging probes. Bioconjugate Chem. 2013, 24, 811–816.
- Kostova, V.; Désos, P.; Starck, J.-B.; Kotschy, A. The chemistry behind ADCs. Pharmaceuticals 2021, 14, 442.
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody–drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881.
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860.
- Abaza, Y.; Fathi, A. Monoclonal Antibodies in Acute Myeloid Leukemia—Are We There Yet? Cancer J. 2022, 28, 37–42.
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821.
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2–positive cancer. Clin. Cancer Res. 2011, 17, 6437–6447.
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46.
- King, H.D.; Dubowchik, G.M.; Mastalerz, H.; Willner, D.; Hofstead, S.J.; Firestone, R.A.; Lasch, S.J.; Trail, P.A. Monoclonal antibody conjugates of doxorubicin prepared with branched peptide linkers: Inhibition of aggregation by methoxytriethyleneglycol chains. J. Med. Chem. 2002, 45, 4336–4343.
- Finbloom, D.S.; Abeles, D.; Rifai, A.; Plotz, P.H. The specificity of uptake of model immune complexes and other protein aggregates by the murine reticuloendothelial system. J. Immunol. 1980, 125, 1060–1065.
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody–drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827.
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735.
- Sunna, A.; Care, A.; Bergquist, P.L. Peptides and Peptide-Based Biomaterials and Their Biomedical Applications; Springer: Berlin/Heidelberg, Germany, 2017.
- Resh, M. Fatty acylation of proteins: The long and the short of it. Prog. Lipid Res. 2016, 63, 120–131.
- Diallo, I.; Seve, M.; Cunin, V.; Minassian, F.; Poisson, J.-F.; Michelland, S.; Bourgoin-Voillard, S. Current trends in protein acetylation analysis. Expert Rev. Proteomic 2019, 16, 139–159.
- Driessen, H.; De Jong, W.; Tesser, G.; Bloemendal, H. The mechanism of N-terminal acetylation of protein. Crit. Rev. Biochem. 1985, 18, 281–325.
- Lee, K.K.; Workman, J. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295.
- Akbarian, M.; Khani, A.; Eghbalpour, S.; Uversky, V.N. Bioactive Peptides: Synthesis, Sources, Applications, and Proposed Mechanisms of Action. Int. J. Mol. Sci. 2022, 23, 1445.
- Silva, O.; Alves, E.S.F.; De La Fuente-Núñez, C.; Ribeiro, S.; Mandal, S.M.; Gaspar, D.; Veiga, A.S.; Castanho, M.; de Andrade, C.A.S.; Nascimento, J.M.; et al. Structural studies of a lipid-binding peptide from tunicate hemocytes with anti-biofilm activity. Sci. Rep. 2016, 6, 27128.
- Zhang, L.; Bulaj, G. Converting peptides into drug leads by lipidation. Curr. Med. Chem. 2012, 19, 1602–1618.
- Grimsey, E.; Collis, D.W.; Mikut, R.; Hilpert, K. The effect of lipidation and glycosylation on short cationic antimicrobial peptides. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 183195.
- Tang, Y.-Q.; Yuan, J.; Osapay, G.; Osapay, K.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated α-defensins. Science 1999, 286, 498–502.
- Craik, D.J.; Daly, N.L.; Bond, T.; Waine, C. Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 1999, 294, 1327–1336.
- Colgrave, M.L.; Craik, D.J. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry 2004, 43, 5965–5975.
- Abdalla, M.A.; McGaw, L.J. Natural cyclic peptides as an attractive modality for therapeutics: A mini review. Molecules 2018, 23, 2080.
- Claro, B.; Bastos, M.; Garcia-Fandino, R. Design and applications of cyclic peptides. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Elsevier: Amsterdam, The Netherlands, 2018; pp. 87–129.
- Kianpour, M.; Akbarian, M.; Uversky, V.N. Nanoparticles for Coronavirus Control. Nanomaterials 2022, 12, 1602.
- Albanese, A.; Tang, P.S.; Chan, W.C. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16.
- Wisse, E.; Braet, F.; Luo, D.; De Zanger, R.; Jans, D.; Crabbe, E.; Vermoesen, A. Structure and function of sinusoidal lining cells in the liver. Toxicol. Pathol. 1996, 24, 100–111.
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756.
- Batista, P.; Castro, P.M.; Madureira, A.R.; Sarmento, B.; Pintado, M. Recent insights in the use of nanocarriers for the oral delivery of bioactive proteins and peptides. Peptides 2018, 101, 112–123.
- Rayaprolu, B.M.; Strawser, J.J.; Anyarambhatla, G. Excipients in parenteral formulations: Selection considerations and effective utilization with small molecules and biologics. Drug Dev. Ind. Pharm. 2018, 44, 1565–1571.
- Rubin, J.; Linden, L.; Coco, W.M.; Bommarius, A.S.; Behrens, S. Salt-induced aggregation of a monoclonal human immunoglobulin G1. J. Pharm. Sci. 2013, 102, 377–386.
- Zhou, P.; Guo, M.; Liu, D.; Liu, X.; Labuza, T. Maillard-reaction-induced modification and aggregation of proteins and hardening of texture in protein bar model systems. J. Food Sci. 2013, 78, C437–C444.
- Joshi, O.; McGuire, J.; Wang, D. Adsorption and function of recombinant factor VIII at solid–water interfaces in the presence of Tween-80. J. Pharm. Sci. 2008, 97, 4741–4755.
- Chi, E.Y.; Weickmann, J.; Carpenter, J.F.; Manning, M.C.; Randolph, T.W. Heterogeneous nucleation-controlled particulate formation of recombinant human platelet-activating factor acetylhydrolase in pharmaceutical formulation. J. Pharm. Sci. 2005, 94, 256–274.
- Allmendinger, A.; Lebouc, V.; Bonati, L.; Woehr, A.; Kishore, R.S.; Abstiens, K. Glass leachables as a nucleation factor for free fatty acid particle formation in biopharmaceutical formulations. J. Pharm. Sci. 2021, 110, 785–795.
- Sharma, B. Immunogenicity of therapeutic proteins. Part 2: Impact of container closures. Biotechnol. Adv. 2007, 25, 318–324.
- Yoneda, S.; Torisu, T.; Uchiyama, S. Development of syringes and vials for delivery of biologics: Current challenges and innovative solutions. Expert Opin. Drug Deliv. 2021, 18, 459–470.
- Srinivasan, C.; Ma, Y.; Liu, Y.; Wang, Y.; Hengst, L.; Liu, X.; Toth, R.; Rodriguez, J.; Mohammad, A.; Bandaranayake, B.M.; et al. Quality attributes and evaluation of pharmaceutical glass containers for parenterals. Int. J. Pharm. 2019, 568, 118510.
- Ditter, D.; Nieto, A.; Mahler, H.-C.; Roehl, H.; Wahl, M.; Huwyler, J.; Allmendinger, A. Evaluation of glass delamination risk in pharmaceutical 10 mL/10R vials. J. Pharm. Sci. 2018, 107, 624–637.
- Jenke, D.R.; Jene, J.M.; Poss, M.; Story, J.; Tsilipetros, T.; Odufu, A.; Terbush, W. Accumulation of extractables in buffer solutions from a polyolefin plastic container. Int. J. Pharm. 2005, 297, 120–133.
- Stan, M.S.; Cinteză, L.O.; Petrescu, L.; Mernea, M.A.; Călborean, O.; Mihailescu, D.F.; Sima, C.; Dinischiotu, A. Dynamic analysis of the interactions between Si/SiO2 quantum dots and biomolecules for improving applications based on nano-bio interfaces. Sci. Rep. 2018, 8, 5289.
- Buchman, J.T.; Elmer, W.H.; Ma, C.; Landy, K.M.; White, J.C.; Haynes, C.L. Chitosan-Coated mesoporous silica nanoparticle treatment of Citrullus lanatus (watermelon): Enhanced fungal disease suppression and modulated expression of stress-related genes. ACS Sustain. Chem. Eng. 2019, 7, 19649–19659.


