 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kazumasa Wakamatsu | -- | 2803 | 2022-11-25 12:47:17 | | | |
| 2 | Jessie Wu | Meta information modification | 2803 | 2022-11-28 04:19:44 | | | | |
| 3 | Jessie Wu | -9 word(s) | 2794 | 2022-11-28 04:28:51 | | |
Video Upload Options
Malignant melanoma is one of the most malignant of all cancers. Melanoma occurs at the epidermo–dermal interface of the skin and mucosa, where small vessels and lymphatics are abundant. Consequently, from the onset of the disease, melanoma easily metastasizes to other organs throughout the body via lymphatic and blood circulation. The most effective treatment method is surgical resection, and other attempted methods, such as chemotherapy, radiotherapy, immunotherapy, targeted therapy, and gene therapy, have not yet produced sufficient results. Since melanogenesis is a unique biochemical pathway that functions only in melanocytes and their neoplastic counterparts, melanoma cells, the development of drugs that target melanogenesis is a promising area of research. Melanin consists of small-molecule derivatives that are always synthesized by melanoma cells. Amelanosis reflects the macroscopic visibility of color changes (hypomelanosis). Under microscopy, melanin pigments and their precursors are present in amelanotic melanoma cells. Tumors can be easily targeted by small molecules that chemically mimic melanogenic substrates. Small-molecule melanin metabolites are toxic to melanocytes and melanoma cells and can kill them.
1. Introduction
2. Chemotherapeutic Approaches Using the Initial Step of Melanogenesis: Tyrosine, Dopa and Their Analogues
- (a)
-
Tyrosine
| Patient no. | Duration of Treatment (hrs) # | Plasma Level (×10−5 M) | Tumor (Percent Labeling Index) |
|
|---|---|---|---|---|
| Preinfusion | Postinfusion | |||
| 1 | 120 | 4.0 | 2.0 | 0.2 |
| 2 | 72 | 5.2 | 3.0 | 0.2 |
| 3 | 48 | 3.5 | 1.0 | 0.1 |
| 4 | 48 | 3.1 | 3.0 | 0.2 |
3. Mechanism of Anti-Melanoma Action in Relation to the Melanogenesis Cascade
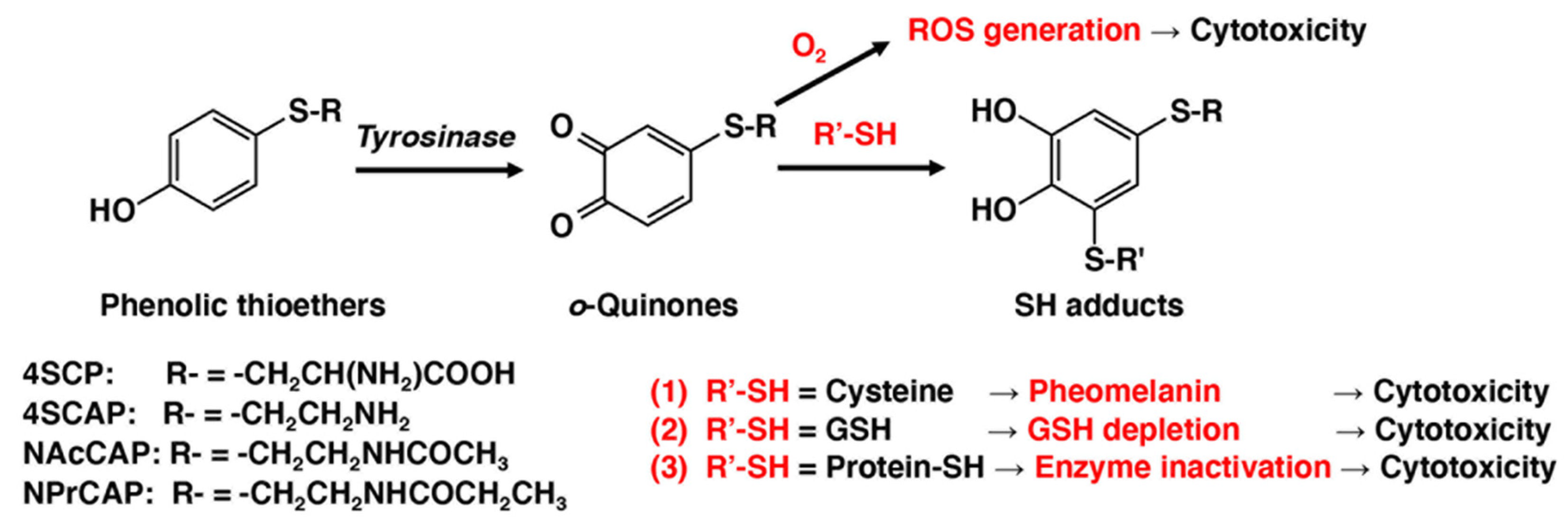
References
- Jimbow, K.; Fitzpatrick, T.B.; Quevedo, W.C., Jr. Formation, chemical compositions and functions of melanin pigments in mammals. Biol. Integument 1986, 2, 278–292.
- Wick, M. An experimental approach to the chemotherapy of melanoma. J. Investig. Dermatol. 1980, 74, 63–65.
- Graham, D.G.; Tiffany, S.M.; Vogel, F.S. The toxicity of melanin precursors. J. Investig. Dermatol. 1978, 70, 113–116.
- Jimbow, K.; Iwashina, T.; Alena, F.; Yamada, K.; Pankovich, J.; Umemura, T. Exploitation of pigment biosynthesis pathway as a selective chemotherapeutic approach for malignant melanoma. J. Investig. Dermatol. 1993, 100, 231S–238S.
- Prota, G. Melanins and Melanogenesis; Academic Press: San Diego, CA, USA, 1992; pp. 1–290.
- Prota, G.; d’Ischia, M.; Mascagna, D. Melanogenesis as a targeting strategy against metastatic melanoma: A reassessment. Melanoma Res. 1994, 4, 351–358.
- Riley, P.A.; Cooksey, C.J.; Johnson, C.I.; Land, E.J.; Latter, A.M.; Ramsden, C.A. Melanogenesis-targeted anti-melanoma pro-drug development: Effect of side chain variations on the cytotoxicity of tyrosinase-generated ortho-quinones in a model screening system. Eur. J. Cancer 1997, 33, 135–143.
- Ito, S.; Sugumaran, M.; Wakamatsu, K. Chemical reactivities of ortho-quinones produced in living organisms: Fate of quinonoid products formed by tyrosinase and phenoloxidase action on phenols and catechols. Int. J. Mol. Sci. 2020, 21, 6080.
- Pawelek, J.M.; Lerner, A.B. 5,6-Dihydroxyindole is a melanin precursor showing potent cytotoxicity. Nature 1978, 276, 626–628.
- Kreider, J.W.; Wade, D.R.; Rosenthal, M.; Densley, T. Maturation and differentiation of B16 melanoma cells induced by theophylline treatment. J. Natl. Cancer Inst. 1975, 54, 1457–1467.
- Pawelek, J.; Wong, G.; Sansone, M.; Morowitz, J. Molecular biology of pigment cells. Molecular controls in mammalian pigmentation. Yale J. Biol. Med. 1973, 46, 430–443.
- Wick, M.M.; Kramer, R.A.; Gorman, M. Enhancement of L-dopa incorporation into melanoma by dopa decarboxylase inhibition. J. Investig. Dermatol. 1978, 70, 358–360.
- Wick, M.M.; Frei, E., 3rd. Selective incorporation of L-3,4-dihydroxyphenylalanine by S-91 Cloudman melanoma in vitro. Cancer Res. 1977, 37, 2123–2125.
- Wick, M.M. L-Dopa methyl ester as a new antitumour agent. Nature 1977, 269, 512–513.
- Wick, M.M. Dopamine: A novel antitumor agent active against B-16 melanoma in vivo. J. Investig. Dermatol. 1978, 71, 163–164.
- Wick, M.M. The chemotherapy of malignant melanoma. J. Investig. Dermatol. 1983, 80 (Suppl. S1), 61s–62s.
- Wick, M.M.; Byers, L.; Ratliff, J. Selective toxicity of 6-hydroxydopa for melanoma cells. J. Investig. Dermatol. 1977, 72, 67–69.
- Fujita, K.; Ito, S.; Inoue, S.; Yamamoto, Y.; Takeuchi, J.; Shamoto, M.; Nagatsu, T. Selective toxicity of 5-S-cysteinyldopa, a melanin precursor, to tumor cells in vitro and in vivo. Cancer Res. 1980, 40, 2543–2546.
- Ito, S.; Inoue, S.; Yamamoto, Y.; Fujita, K. Synthesis and antitumor activity of cysteinyl-3,4-dihydroxyphenylalanines and related compounds. J. Med. Chem. 1981, 24, 673–677.
- Graham, D.G.; Tiffany, S.M.; Bell, W.R., Jr.; Gutknecht, W.F. Autoxidation versus covalent binding of quinones as the mechanism of toxicity of dopamine, 6-hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol. Pharmacol. 1978, 14, 644–653.
- Ito, S.; Inoue, S.; Fujita, K. The mechanism of toxicity of 5-S-cysteinyldopa to tumour cells. Hydrogen peroxide as a mediator of cytotoxicity. Biochem. Pharmacol. 1983, 32, 2079–2081.
- Jimbow, K.; Miura, T.; Ito, S.; Ishikawa, K. Phenolic melanin precursors provide a rational approach to the design of antitumor agents for melanoma. Pigment Cell Res. 1989, 2, 34–39.
- Pankovich, J.M.; Jimbow, K. Tyrosine transport in a human melanoma cell line as a basis for selective transport of cytotoxic analogues. Biochem. J. 1991, 280, 721–725.
- Nakamura, T.; Seki, S.; Matsubara, O.; Ito, S.; Kasuga, T. Specific incorporation of 4-S-cysteinylphenol into human melanoma cells. J. Investig. Dermatol. 1988, 90, 725–728.
- Yamada, I.; Seki, S.; Matsubara, O.; Ito, S.; Suzuki, S.; Kasuga, T. The cytotoxicity of cysteinylcatechols and related compounds to human melanoma cells in vitro. J. Investig. Dermatol. 1987, 88, 538–540.
- Miura, S.; Ueda, T.; Jimbow, K.; Ito, S.; Fujita, K. Synthesis of cysteinylphenol, cysteaminylphenol and related compounds and in vivo evaluation of anti-melanoma effect. Arch. Dermatol. Res. 1987, 279, 219–225.
- Ito, Y.; Jimbow, K. Selective cytotoxicity of 4-S-cysteaminylphenol on follicular melanocytes of the black mouse: Rational basis for its application to melanoma chemotherapy. Cancer Res. 1987, 47, 3278–3284.
- Kitagawa, M.; Nemoto, T.; Seki, S.; Ito, S.; Kasuga, T. In vivo antimelanoma effects of 4-S-cysteaminylphenol, a newly synthesized therapeutic agent specific to melanoma. J. Cancer Res. Clin. Oncol. 1993, 119, 470–474.
- Ito, Y.; Jimbow, K.; Ito, S. Depigmentation of black guinea pig skin by topical application of cysteaminylphenol, cysteinylphenol, and related compounds. J. Investig. Dermatol. 1987, 88, 77–82.
- Alena, F.; Jimbow, K.; Ito, S. Melanocytotoxicity and antimelanoma effects of phenolic amine compounds in mice in vivo. Cancer Res. 1990, 50, 3743–3747.
- Miura, T.; Jimbow, K.; Ito, S. The in vivo antimelanoma effect of 4-S-cysteaminylphenol and its N-acetyl derivative. Int. J. Cancer 1990, 46, 931–934.
- Padgette, S.R.; Herman, H.H.; Han, J.H.; Pollock, S.H.; May, S.W. Antihypertensive activities of phenyl aminoethyl sulfides, a class of synthetic substrates for dopamine hydroxylase. J. Med. Chem. 1984, 27, 1354–1357.
- Pankovich, J.M.; Jimbow, K.; Ito, S. 4-S-cysteaminylphenol and its analogues as substrates for tyrosinase and monoamine oxidase. Pigment Cell Res. 1990, 3, 146–149.
- Liu, W.-Y.; Lin, C.-C.; Hsieh, Y.-S.; Wu, Y.-T. Nanoformulation development to improve the biopharmaceutical properties of fisetin using design of experiment approach. Molecules 2021, 26, 3031.
- Inoue, S.; Ito, S.; Wakamatsu, K.; Jimbow, K.; Fujita, K. Mechanism of growth inhibition of melanoma cells by 4-S-cysteaminylphenol and its analogues. Biochem. Pharmacol. 1990, 39, 1077–1083.
- Yukitake, J.; Otake, H.; Inoue, S.; Wakamatsu, K.; Olivares, C.; Solano, F.; Hasegawa, K.; Ito, S. Synthesis and selective in vitro anti-melanoma effect of enantiomeric alpha-methyl- and alpha-ethyl-4-S-cysteaminylphenol. Melanoma Res. 2003, 13, 603–609.
- Yukitake, J.; Otake, H.; Inoue, S.; Wakamatsu, K.; Ito, S. Comparison of in vivo anti-melanoma effect of enantiomeric alpha-methyl- and alpha-ethyl-4-S-cysteaminylphenol. Melanoma Res. 2004, 14, 116–120.
- Inoue, S.; Hasegawa, K.; Ito, S.; Ozeki, H.; Solano, F.; Jiménez-Cervantes, C.; Wakamatsu, K.; Fujita, K. Antimelanoma effect of 4-S-cysteaminylcatechol, an activated form of 4-S-cysteaminylphenol. Cancer Res. 1995, 55, 2603–2607.
- Wong, M.; Jimbow, K. Selective cytotoxicity of N-acetyl-4-S-cysteaminylphenol on follicular melanocytes of black mice. Brit. J. Dermatol. 1991, 124, 56–61.
- Thomas, P.D.; Kishi, H.; Cao, H.; Ota, M.; Yamashita, T.; Singh, S.; Jimbow, K. Selective incorporation and specific cytocidal effect as the cellular basis for the antimelanoma action of sulphur containing tyrosine analogs. J. Investig. Dermatol. 1999, 113, 928–934.
- Tandon, M.; Thomas, P.D.; Shokravi, M.; Singh, S.; Samra, S.; Chang, D.; Jimbow, K. Synthesis and antitumor effect of the melanogenesis-based antimelanoma agent N-propionyl-4-S-cysteaminylphenol. Biochem. Pharmacol. 1998, 55, 2023–2029.
- Minamitsuji, Y.; Toyofuku, K.; Sugiyama, S.; Yamada, K.; Jimbow, K. Sulfur containing tyrosine analogs can cause selective melanocytotoxocity involving tyrosinase-mediated apoptosis. J. Investig. Dermatol. Symp. Proc. 1999, 4, 130–136.
- Lant, N.J.; McKeown, P.; Kelland, L.R.; Rogers, P.M.; Robins, D.J. Synthesis and antimelanoma activity of analogues of N-acetyl-4-S-cysteaminylphenol. Anticancer. Drug Des. 2000, 15, 295–302.
- Lant, N.J.; McKeown, P.; Timoney, M.C.; Kelland, L.R.; Rogers, P.M.; Robins, D.J. Synthesis and anti-melanoma activity of analogues of N-acetyl-4-S-cysteaminylphenol substituted with two methyl groups alpha to the nitrogen. Anticancer. Drug Des. 2001, 6, 49–55.
- Pearson, V.C.; Ferguson, J.; Rogers, P.M.; Kelland, L.R.; Robins, D.J. Synthesis and antimelanoma activity of tertiary amide analogues of N-acetyl-4-S-cysteaminylphenol. Oncol. Res. 2003, 13, 503–512.
- Ferguson, J.; Rogers, P.M.; Kelland, L.R.; Robins, D.J. Synthesis and antimelanoma activity of sterically congested tertiary amide analogues of N-acetyl-4-S-cysteaminylphenol. Oncol. Res. 2005, 15, 87–94.
- Nicoll, K.; Robertson, J.; Lant, N.; Kelland, L.R.; Rogers, P.M.; Robins, D.J. Synthesis and antimelanoma activity of reversed amide analogues of N-acetyl-4-S-cysteaminylphenol. Oncol. Res. 2006, 16, 97–106.
- Granada, M.; Mendes, E.; Perry, M.J.; Penetra, M.J.; Gaspar, M.M.; Pinho, J.O.; Serra, S.; António, C.T.; Francisco, A.P. Sulfur Analogues of Tyrosine in the Development of Triazene Hybrid Compounds: A New Strategy against Melanoma. ACS Med. Chem. Lett. 2021, 12, 1669–1677.
- Yamada, I.; Seki, S.; Ito, S.; Suzuki, S.; Matsubara, O.; Kasuga, T. The killing effect of 4-S-cysteaminylphenol, a newly synthesised melanin precursor, on B16 melanoma cell lines. Br. J. Cancer 1991, 63, 187–190.
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395.
- Tse, D.C.; McCreery, R.L.; Adams, R.N. Potential oxidative pathways of brain catecholamines. J. Med. Chem. 1976, 19, 37–40.
- Tanaka, H.; Yamashita, Y.; Umezawa, K.; Hirobe, T.; Ito, S.; Wakamatsu, K. The pro-oxidant activity of pheomelanin is significantly enhanced by UVA irradiation: Benzothiazole moieties are more reactive than benzothiazine moieties. Int. J. Mol. Sci. 2018, 19, 2889.
- Alena, F.; Dixon, W.; Thomas, P.; Jimbow, K. Glutathione plays a key role in the depigmenting and melanocytotoxic action of N-acetyl-4-S-cysteaminylphenol in black and yellow hair follicles. J. Investig. Dermatol. 1995, 104, 792–797.
- Alena, F.; Iwashina, T.; Gili, A.; Jimbow, K. Selective in vivo accumulation of N-acetyl-4-S-cysteaminylphenol in B16F10 murine melanoma and enhancement of its in vitro and in vivo antimelanoma effect by combination of buthionine sulfoximine. Cancer Res. 1994, 54, 2661–2666.
- Ito, S.; Kato, T.; Fujita, K. Covalent binding of catechols to proteins through the sulphydryl group. Biochem. Pharmacol. 1988, 37, 1707–1710.
- Prezioso, J.A.; Wang, N.; Bloomer, W.D. Thymidylate synthase as a target enzyme for the melanoma-specific toxicity of 4-S-cysteaminylphenol and N-acetyl-4-S-cysteaminylphenol. Cancer Chemother. Pharmacol. 1992, 30, 394–400.
- Ito, S.; Kato, T.; Ishikawa, K.; Kasuga, T.; Jimbow, K. Mechanism of selective toxicity of 4-S-cysteinylphenol and 4-S-cysteaminylphenol to melanocytes. Biochem. Pharmacol. 1987, 36, 2007–2011.
- Penning, T.M. Genotoxicity of ortho-quinones: Reactive oxygen species versus covalent modification. Toxicol. Res. 2017, 6, 740–754.
- Dunsmore, L.; Navo, C.D.; Becher, J.; de Montes, E.G.; Guerreiro, A.; Hoyt, E.; Brown, L.; Zelenay, V.; Mikutis, S.; Copper, J.; et al. Controlled masking and targeted release of redox-cycling ortho-quinones via a C-C bond-cleaving 1,6-elimination. Nat. Chem. 2022, 14, 754–765.
- Gutierrez, P.L. The metabolism of quinone-containing alkylating agents: Free radical production and measurement. Front. Biosci. 2000, 5, D629–D638.
- Mascagna, D.; Costantini, C.; d’Ischia, M.; Prota, G. Biomimetic oxidation of the antimelanoma agent 4-S-cysteaminylphenol and related catechol thioethers: Isolation and reaction behaviour of novel dihydrobenzothiazinequinones. Tetrahedron 1994, 50, 8757–8764.
- Hasegawa, K.; Ito, S.; Inoue, S.; Wakamatsu, K.; Ozeki, H.; Ishiguro, I. Dihydro-1,4-benzothiazine-6,7-dione, the ultimate toxic metabolite of 4-S-cysteaminylphenol and 4-S-cysteaminylcatechol. Biochem. Pharmacol. 1997, 53, 1435–1444.
- Ishii-Osai, Y.; Yamashita, T.; Tamura, Y.; Sato, N.; Ito, A.; Honda, H.; Wakamatsu, K.; Ito, S.; Nakayama, E.; Okura, M.; et al. N-propionyl-4-S-cysteaminylphenol induces apoptosis in B16F1 cells and mediates tumor-specific T-cell immune responses in a mouse melanoma model. J. Dermatol. Sci. 2012, 67, 51–60.
- Ito, S.; Nishigaki, A.; Ishii-Osai, Y.; Ojika, M.; Wakamatsu, K.; Yamashita, T.; Tamura, Y.; Ito, A.; Honda, H.; Nakayama, E.; et al. Mechanism of putative neo-antigen formation from N-propionyl-4-S-cysteaminylphenol, a tyrosinase substrate, in melanoma models. Biochem. Pharmacol. 2012, 84, 646–653.

