1. Introduction
Malignant melanoma possesses a unique metabolic pathway, the synthesis of melanin pigments, which are formed by the conversion of tyrosine to dopaquinone in the presence of tyrosinase (EC 1.14.18.1). This process occurs in specific secretory granules: melanosomes [
1]. Different synthetic stages of melanin pigments have been exploited to develop melanoma cell-specific chemotherapeutic agents [
2]. Most previous attempts have utilized dopa and related catechol compounds, which cause general cytotoxicity through autoxidation [
3].
Tyrosine analogues, which are tyrosinase substrates, are, however, excellent candidates for melanoma-specific targeting therapy [
4]. Melanogenesis, a biochemical process unique to melanocytes; is highly expressed in malignant melanoma. A specific enzyme tyrosinase catalyzes the oxidative conversion of L-tyrosine to melanin pigments in melanocytes and malignant melanoma cells [
5]. Harnessing melanogenesis to develop melanoma-specific antitumor agents has been a challenging goal [
6]. Tyrosinase can oxidize a variety of natural and synthetic phenols to produce highly reactive and cytotoxic
o-quinones [
7,
8].
2. Chemotherapeutic Approaches Using the Initial Step of Melanogenesis: Tyrosine, Dopa and Their Analogues
There are two major categories of tyrosinase-interacting melanin precursors in the initial step of melanin biosynthesis that were examined as potential sources when designing antimelanoma agents. They were tyrosine (monohydroxy = phenolic) and dopa (L-dopa, levodopa) (dihydroxy = catecholic) [
2]. 5,6-Dihydroxyindole is another melanogenesis-related group that has been explored for developing tyrosinase-mediated antimelanoma agents [
49]. It is, however, extremely reactive, producing reactive oxygen species (ROS), and this reactivity has limited dihydroxyindoles’ role as antitumor agents.
- (a)
-
Tyrosine
The degree of melanin pigmentation of in vitro melanoma cells can be altered by changing the concentration of tyrosine in the culture medium [
50]. Raising the concentration of tyrosine causes an increase in vitro melanoma cell growth. Tyrosine levels may be elevated enough to cause toxicity to melanin-forming cells [
51]. This technique has proved to be a biologically useful method. A major obstacle to applying this approach for melanoma therapy is, however, that tyrosine is one of the naturally occurring amino acids and is extensively used in general protein synthesis. Rapidly growing melanocytes incorporate only about 5% of exogenous tyrosine into melanin biosynthesis, with the remainder entering protein synthesis. Therapeutic approaches to raise or lower levels of tyrosine in vivo have, therefore, had limited success.
L-dopa is an amino acid, but it is not normally found in cellular proteins. L-dopa may, therefore, be selectively incorporated into melanoma cells. Through the use of L-dopa decarboxylase inhibitors, which limit the decarboxylation of L-dopa to dopamine, an enhanced incorporation of L-dopa into melanoma tissues was achieved in vivo [
52]. L-dopa was selectively cytotoxic to melanoma cells in vitro [
53], and through the use of its more water-soluble analog L-dopa methyl ester, in vivo antimelanoma activity was observed in the experimental murine melanoma [
54]. In this animal experiment, a hypercatecholamine-type state was observed [
54]. The animals became agitated and tremulous and usually died within 1–2 h after administration. These acute toxic effects of L-dopa are probably mediated by its conversion to dopamine by the enzyme, dopadecarboxylase. Furthermore, the major metabolite of L-dopa, dopamine, was observed to be a highly potent inhibitor of melanoma cells in vivo and in vitro [
55]. Interestingly, however, four patients with extensive melanoma metastases were treated by dopamine infusions at maximal tolerable levels for from 48 to 120 h and labeling and scintillation indices of the tumors were measured. A comparison of the pre- and post-infusion data showed a significant reduction in the % labelling index, from 1.0~3.0 to 0.1~0.2, indicating a consistent 10-fold reduction. However, persistent fatigue precluded further retreatment [
56] (
Table 1).
One of the L-dopa analogues, 6-hydroxydopa, was found to be highly potent in terms of selective interaction with tyrosinase and toxicity for melanoma cells [
49,
57]. However, in in vivo experiments, 6-hydroxydopa was found to exhibit unique neurotoxic properties that resulted in the selective degeneration of adrenergic nerves [
2].
Following those efforts by Wick’s group, Fujita et al. reported that 5-
S-cysteinyldopa (5SCD), a pheomelanin precursor, was selectively toxic to melanoma cells in vitro and in vivo [
58]. This study showed that 1 mM 5SCD exhibited a similar or an even higher degree of growth inhibition to 6 mM L-dopa. In an extension of this study, Ito et al. synthesized several new molecules that are structurally related to 5SCD and demonstrated melanoma activity in vitro and in vivo [
9]. However, the use of catecholic melanin precursors such as L-dopa and 5SCD faced a drawback in that they tend to undergo autoxidation [
59] leading to the production of reactive oxygen species such as hydrogen peroxide [
60], resulting in non-selective cytotoxicity [
4,
9].
The use of phenolic melanin precursors appeared more rational and promising for the development of melanogenesis-based anti-melanoma agents because the oxidation of phenolic compounds is only dependent on tyrosinase present in melanocytes and melanoma cells. To examine this possibility, 4-
S-cysteinylphenol (4SCP), a phenolic thioether, was synthetized by heating phenol and cystine in hydrobromic acid, and its anti-melanoma effects were evaluated [
9]. 4SCP is the sulfur homolog of L-tyrosine, the natural melanin precursor, and was expected to be a good substrate for tyrosinase. In fact, 4SCP was found to be as good a substrate for melanoma tyrosinase as L-tyrosine [
16] and a good inhibitor of L-tyrosine transport to melanoma cells, implying the efficient transportation of 4SCP [
61], and
3H-4SCP was specifically incorporated into cultured human melanoma cells in vitro [
62] and mouse melanoma tissues in vivo [
16]. However, the antimelanoma effects of 4SCP were found to be minimal [
16,
21], which led us to modify its structure to the amine analog 4-
S-cysteaminylphenol (4SCAP), in the hope that this modification would lead to the more effective incorporation into melanoma cells and oxidation by tyrosinase to elicit greater antimelanoma effects. 4SCAP was readily prepared by heating phenol and cystamine in hydrobromic acid [
10]. In fact, 4SCAP was found to cause a significant inhibition of in vivo melanoma growth and marked depigmentation of black skin and hair follicles, with the effects being much greater than those caused by 4SCP [
10,
23,
25,
28,
29]. When hair follicles were plucked from adult black mice to stimulate new melanocyte growth and activate tyrosinase synthesis, subsequent repeated intraperitoneal injection (ip) administration of 4SCAP resulted in 100% new growth of white hair follicles at the site at which black follicles were plucked [
23]. 4SCAP also suppressed lung colony formation after intravenous inoculation of B16 melanoma cells in mice [
25,
29]. These promising in vivo results were paralleled by in vitro studies showing that 4SCAP was much more toxic to cultured human melanoma cells than 4SCP [
21] and more potent in inhibiting protein synthesis [
16].
However, there were certain drawbacks to further exploiting 4SCAP as an antimelanoma agent: the maximal tolerable dose was limited due to its low water solubility, low LD
50 value [
24], and hypotensive effect [
14]. 4SCAP acts as a substrate for a number of enzymes in addition to tyrosinase, including dopamine β-hydroxylase [
14] and monoamine oxidase [
17], which catalyze production of the sulfoxide and aldehyde, respectively. As a result, 4SCAP appeared to cause adverse effects when administrated systematically. Recent progress in nanotechnology may overcome the limitations, such as low solubility and adverse effects of compounds, and exploring the nanoformulation of 4SCAP is a possible approach [
63]. Alternatively, we took the approach of modifying 4SCAP by chemical synthesis. 4SCAP homologues such as α-methyl-4SCAP, 4-
S-homocysteaminylphenol, and
N,
N-dimethyl-4SCAP were also prepared, and their depigmenting effects were examined in vitro [
11] and in vivo [
29]. However, none were found to be superior to 4SCAP. Attempts to lower the hypotensive effect of 4SCAP and increase the efficacy of tyrosinase-dependent cytotoxicity were made using enantiomers of α-methyl-4SCAP and α-ethyl-4SCAP [
12,
26]. However, improvements were limited. Another attempt to increase the efficacy of 4SCAP as an antimelanoma agent, using the catecholic derivative 4-
S-cysteaminylcatechol (4SCAC), had limited success due to systemic toxicity [
13]. To overcome these difficulties, we prepared
N-acetyl-4SCAP (NAcCAP) by reacting 4-hydroxythiophenol with 2-methyl-2-oxazoline using the method of Padgette et al. (1984) [
14].
NAcCAP was found to act as a substrate for mushroom tyrosinase as well as 4SCAP, and exhibit greater in vivo antimelanoma activity than 4SCAP, although at higher doses [
24]. Importantly, NAcCAP showed marked water solubility [
24] and a greater depigmenting effect on follicular melanocytes than 4SCAP [
29]. A single ip administration of NAcCAP into newborn mice led to the development of silver hair follicles in the entire body. The selective destruction of melanocytes was observed at 12 hr after a single ip injection. None of the surrounding keratinocytes or fibroblasts showed such subcellular degeneration and cell death [
30].
14C-NAcCAP was specifically taken up by melanotic melanoma cells, but not by amelanotic melanoma cells [
19].
These promising results that were achieved by protecting the amino group in 4SCAP were elaborated further by replacing the
N-acetyl group with an
N-propionyl group. NPrCAP was readily prepared by reacting 4-hydroxythiophenol with 2-ethyl-2-oxazoline [
15]. NPrCAP was expected to exhibit greater uptake into melanoma cells because of its greater lipophilicity compared with NAcCAP. NPrCAP was found to have a greater Vmax value for tyrosinase than tyrosine, 4SCAP, and NAcCAP had, and caused marked depigmentation of black hair follicles in adult and newborn C57 mice, with biochemical and morphologic features indicative of apoptosis [
15,
18]. The tyrosinase-mediated cytotoxicity of NPrCAP was further confirmed by the finding of decreased viability of tyrosinase-transfected COS7 monkey-kidney cells expressing high tyrosinase activity. NPrCAP, however, also transiently inhibited the proliferation of a tyrosinase-negative albino melanocyte line, as well as COS7 cells [
18]. It appears that the major process of NPrCAP melanocyte-toxicity involves cytocidal apoptosis associated with active tyrosinase. In addition, there is transient, non-tyrosinase-mediated cytostatic cytotoxicity. Antimelanoma effects were also compared for NPrCAP and NAcCAP in B16 melanoma tumors; the two compounds were comparable in terms of their growth-inhibitory effect, but NPrCAP was considerably better than NAcCAP in increasing the lifespan of melanoma-bearing mice [
15]. Taken together, NPrCAP appears to be the best antimelanoma agent among the sulfur-containing tyrosine analogs (phenolic thioethers) developed to date.
A number of attempts to increase the efficacy of NAcCAP have been reported. Robins et al. [
64,
65,
66,
67,
68] synthesized various NAcCAP analogues with the intention of increasing the lipophilicity of the compounds. A modest increase in antimelanoma activity against several melanoma cell lines was observed, which was correlated with increased lipophilicity. However, those compounds also exhibited tyrosinase-independent cytotoxicity against an amelanotic SK-Mel-24 melanoma and an ovarian cell line. Of particular interest is a recent report showing a hybrid of 4SCAP with triazene, a DNA-alkylating compound [
69]. Those hybrids were found to be excellent tyrosinase substrates. Some of those compounds were unexpectedly devoid of hepatotoxicity while maintaining cytotoxic activity in melanoma cells. 4SCAP appears to be an important component for the new strategy of developing anti-melanoma agents.
3. Mechanism of Anti-Melanoma Action in Relation to the Melanogenesis Cascade
Here, we describe the possible mechanisms of anti-melanoma action of 4SCAP, NAcCAP/NPrCAP, and related phenols. Tyrosinase is certainly a trigger of anti-melanoma effects of 4SCAP based on the results that (1) 2-
S-cysteaminylphenol, an isomer of 4SCAP, did not act as a substrate of tyrosinase and did not exhibit any anti-melanoma and depigmenting effects [
10], (2) the cytotoxicity of 4SCAP against various melanoma cell lines depended on the degree of their pigmentation [
22], and (3) 4SCAP exhibited cytotoxicity to melanocytes in black mice but not in albino mice [
23].
Tyrosinase is known to catalyze oxidation of various phenols to the corresponding
o-quinones [
70].
o-Quinones are highly reactive molecules through binding with various functional groups, especially sulfhydryl and amino groups [
8,
71]. The binding of
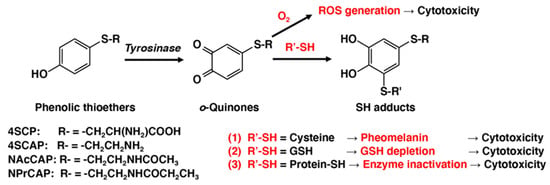
o-quinones with the cellular small sulfhydryl compounds cysteine and glutathione (GSH) produces the cysteinyl- and glutathionyl-catechol derivatives. This process results in various biochemical consequences in melanocytes and melanoma cells (
Figure 1). One is the production of pheomelanic pigments by the oxidation of cysteinyl-catechol conjugates, similar to the production of natural pheomelanin from L-tyrosine. Pheomelanins are known to exhibit potent pro-oxidant activities [
72]. Another is the depletion of GSH in cells that exhibit tyrosinase activity. Both of these biochemical events might lead to tyrosinase-dependent cytotoxicity. GSH depletion was shown to play a key role in the depigmenting and melanocytic action of NAcCAP: the co-administration of
N-acetylcysteine, which up-regulated GSH content, completely abolished the depigmenting potency of NAcCAP, whereas buthionine sulfoximine, which depleted the tissue GSH content, enhanced the depigmenting potency of NAcCAP [
73]. The co-administration of buthionine sulfoximine also significantly enhanced the antimelanoma effects of NAcCAP [
74]. Additionally,
o-quinones are capable of binding with enzymes and other proteins through their cysteine residues [
75]. The binding of
o-quinones to sulfhydryl enzymes essential for proliferation, such as thymidylate synthase and DNA polymerase, leads to the inhibition of DNA synthesis and cell growth [
76]. Interestingly, upon tyrosinase oxidation, 4SCAP was found to be five-fold more effective than 4SCP in binding to alcohol dehydrogenase, a sulfhydryl enzyme [
16,
77].
Figure 1. Mechanisms of cytotoxicity of o-quinones produced by tyrosinase-catalyzed oxidation of phenolic thioethers.
In addition to the cytotoxicity of
o-quinone due to their reactivity with sulfhydryl compounds, there is another possible mechanism for
o-quinone cytotoxicity (
Figure 1).
o-Quinones may undergo one-electron reduction with reducing agents such as NAD(P)H and Fe
2+ to produce semi-quinone radicals, which disproportionately form the catechol and the
o-quinone [
78,
79]. Catechols are converted back to
o-quinones upon autoxidation, concomitantly producing ROS such as superoxide radical, hydrogen peroxide, and hydroxyl radical.
o-Quinones may also undergo two-electron reduction, with reducing enzymes such as NAD(P)H quinone dehydrogenase 1 (NQO1) or reducing compounds such as ascorbic acid, to produce catechols. Thus, both catechols and
o-quinones can be a source of ROS generation through redox cycling [
78,
79,
80]. In addition, the reactions of
o-quinones with biological small thiols such as cysteine and GSH produce catechol derivatives that may produce ROS. Interestingly, catechols with cysteine conjugation, such as 5SCD, are more cytotoxic to melanocytes through the production of hydrogen peroxide than are the parent catechols such as L-dopa [
58,
60].
Whether a catechol or its corresponding
o-quinone structure is more cytotoxic depends on the reactivity of the catechol: more reactive and, thus, cytotoxic catechols such as 6-hydroxydopamine become less reactive when oxidized to the
o-quinone structure [
59]. On the other hand, the sulfhydryl reactivity of
o-quinone oxidation products contributes to the cytotoxicity of dopamine and
N-acetyldopamine [
59]. In our case, which of the two mechanisms prevails, the sulfhydryl reaction or the ROS production, is not known at present.
A unique feature of 4SCAP is that, upon tyrosinase-catalyzed oxidation,
o-quinone derived from 4SCAP undergoes a facile cyclization through the amino group to form a reactive, cyclic quinone, dihydro-1,4-benzothiazine-6,7-dione [
81,
82]. This cyclic quinone was found to be highly cytotoxic to B16 melanoma cells in vitro and in vivo, much more so than 4SCAP [
82].
Lastly, researchers examined how extensively ROS are involved in the tyrosinase-dependent antimelanoma and depigmenting effects of 4SCAP and NAcCAP (or NPrCAP). The production of ROS from the phenolic tyrosinase substrate NPrCAP in melanoma cells was reported by [
27]. This study showed that the growth suppression of pigmented melanoma cells by NPrCAP was associated with an increase in intracellular ROS, activation of caspase 3, and DNA fragmentation. This study also showed that intratumoral administration of NPrCAP suppressed the growth in not only primary B16F1 melanoma tumors but also secondary, re-challenged tumors. The participation of CD8
+ T cells was suggested for the NPrCAP-mediated anti-B16 melanoma immunity. In a following study, the molecular mechanisms of the NPrCAP cytotoxicity and immunogenicity were examined [
20]. NPrCAP was shown to be oxidized by mushroom tyrosinase to the
o-quinone
N-propionyl-4-
S-cysteaminyl-1,2-benzoquinone (NPrCAQ). NPrCAQ rapidly reacted with biologically relevant thiols, cysteine, GSH, and bovine serum albumin through the sulfhydryl group (
Figure 1). The production and excretion of NPrCAQ-protein adducts was confirmed in B16F1 melanoma cells in vitro and in B16F1 melanoma-bearing mice in vivo. The protein fraction was hydrolyzed by heating in 6 M HCl and the amino acid released from the protein adduct, 5-
S-cysteaminyl-3-
S-cysteinylcatechol, was identified by HPLC. These results suggest that tyrosinase in melanoma cells activate the phenolic NPrCAP, acting as a prohapten, to the quinone-hapten NPrCAQ, which binds to melanosomal proteins to form possible neo-antigens, thus triggering an immunological response.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14225588