 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tatyana Merkulova | -- | 1892 | 2022-11-22 10:04:30 | | | |
| 2 | Sirius Huang | -3 word(s) | 1889 | 2022-11-23 02:35:01 | | | | |
| 3 | Sirius Huang | -1 word(s) | 1888 | 2022-11-24 07:40:25 | | |
Video Upload Options
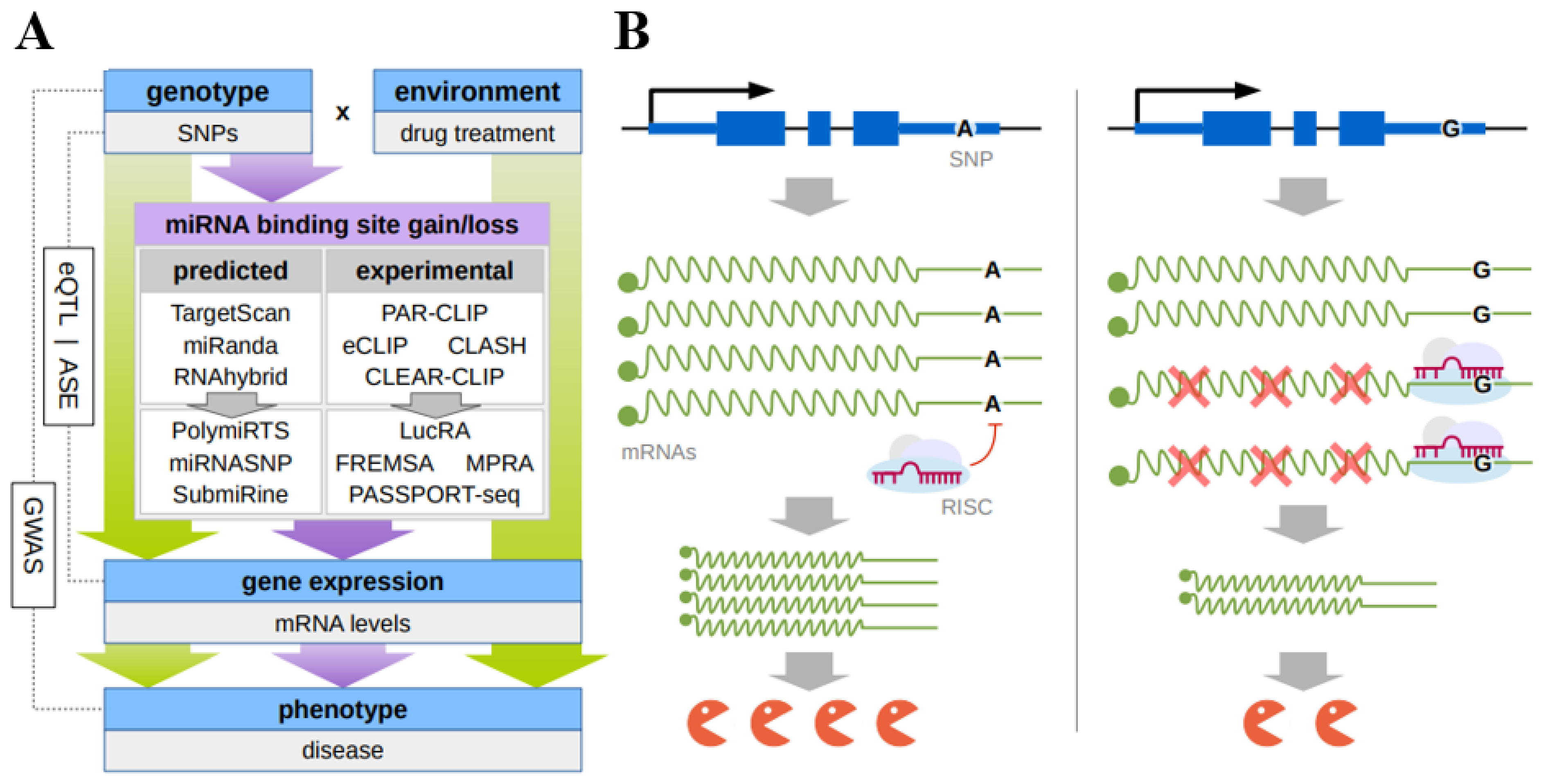
The complementary interaction of microRNAs (miRNAs) with their binding sites in the 3′untranslated regions (3′UTRs) of target gene mRNAs represses translation, playing a leading role in gene expression control. MiRNA recognition elements (MREs) in the 3′UTRs of genes often contain single nucleotide polymorphisms (SNPs), which can change the binding affinity for target miRNAs leading to dysregulated gene expression. Accumulated data suggest that these SNPs can be associated with various human pathologies (cancer, diabetes, neuropsychiatric disorders, and cardiovascular diseases) by disturbing the interaction of miRNAs with their MREs located in mRNA 3′UTRs. Numerous data show the role of SNPs in 3′UTR MREs in individual drug susceptibility and drug resistance mechanisms. This work brief the data on such SNPs focusing on the most rigorously proven cases. Some SNPs belong to conventional genes from the drug-metabolizing system (in particular, the genes coding for cytochromes P450 (CYP 450), phase II enzymes (SULT1A1 and UGT1A), and ABCB3 transporter and their expression regulators (PXR and GATA4)). Other examples of SNPs are related to the genes involved in DNA repair, RNA editing, and specific drug metabolisms. The gene-by-gene studies and genome-wide approaches utilized or potentially utilizable to detect the MRE SNPs associated with individual response to drugs discussed.
1. Introduction
2. Modern Approaches to Identify Functional SNPs in 3′UTR miRNA Target Sequences
References
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and Limitations of Genome-Wide Association Studies. Nat. Rev. Genet. 2019, 20, 467–484.
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of Published Genome-Wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012.
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A Brief History of Human Disease Genetics. Nature 2020, 577, 179–189.
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential Etiologic and Functional Implications of Genome-Wide Association Loci for Human Diseases and Traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367.
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195.
- Huang, Q. Genetic Study of Complex Diseases in the Post-GWAS Era. J. Genet. Genom. 2015, 42, 87–98.
- Degtyareva, A.O.; Antontseva, E.V.; Merkulova, T.I. Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases. Int. J. Mol. Sci. 2021, 22, 6454.
- Jin, Y.; Jiang, J.; Wang, R.; Qin, Z.S. Systematic Evaluation of DNA Sequence Variations on in Vivo Transcription Factor Binding Affinity. Front. Genet. 2021, 12, 667866.
- Tseng, C.-C.; Wong, M.-C.; Liao, W.-T.; Chen, C.-J.; Lee, S.-C.; Yen, J.-H.; Chang, S.-J. Genetic Variants in Transcription Factor Binding Sites in Humans: Triggered by Natural Selection and Triggers of Diseases. Int. J. Mol. Sci. 2021, 22, 4187.
- Dufner-Almeida, L.G.; do Carmo, R.T.; Masotti, C.; Haddad, L.A. Understanding Human DNA Variants Affecting Pre-MRNA Splicing in the NGS Era. Adv. Genet. 2019, 103, 39–90.
- Li, B.; Dong, J.; Yu, J.; Fan, Y.; Shang, L.; Zhou, X.; Bai, Y. Pinpointing MiRNA and Genes Enrichment over Trait-Relevant Tissue Network in Genome-Wide Association Studies. BMC Med. Genom. 2020, 13, 191.
- Chhichholiya, Y.; Suryan, A.K.; Suman, P.; Munshi, A.; Singh, S. SNPs in MiRNAs and Target Sequences: Role in Cancer and Diabetes. Front. Genet. 2021, 12, 793523.
- Moszyńska, A.; Gebert, M.; Collawn, J.F.; Bartoszewski, R. SNPs in MicroRNA Target Sites and Their Potential Role in Human Disease. Open Biol. 2017, 7, 170019.
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of MRNA Translation and Stability by MicroRNAs. Annu. Rev. Biochem. 2010, 79, 351–379.
- Wilczynska, A.; Bushell, M. The Complexity of MiRNA-Mediated Repression. Cell Death. Differ. 2015, 22, 22–33.
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162.
- Nakano, M.; Iwakami, C.; Fukami, T.; Nakajima, M. Identification of MiRNAs That Regulate Human CYP2B6 Expression. Drug Metab. Pharmacokinet. 2021, 38, 100388.
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233.
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. MiRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276.
- He, B.; Zhao, Z.; Cai, Q.; Zhang, Y.; Zhang, P.; Shi, S.; Xie, H.; Peng, X.; Yin, W.; Tao, Y.; et al. MiRNA-Based Biomarkers, Therapies, and Resistance in Cancer. Int. J. Biol. Sci. 2020, 16, 2628–2647.
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.-S. Therapeutic Advances of MiRNAs: A Preclinical and Clinical Update. J. Adv. Res. 2021, 28, 127–138.
- Paul, S.; Reyes, P.R.; Garza, B.S.; Sharma, A. MicroRNAs and Child Neuropsychiatric Disorders: A Brief Review. Neurochem. Res. 2020, 45, 232–240.
- Paul, S.; Bravo Vázquez, L.A.; Uribe, S.P.; Manzanero Cárdenas, L.A.; Ruíz Aguilar, M.F.; Chakraborty, S.; Sharma, A. Roles of MicroRNAs in Carbohydrate and Lipid Metabolism Disorders and Their Therapeutic Potential. Biochimie 2021, 187, 83–93.
- Swart, M.; Dandara, C. Genetic Variation in the 3’-UTR of CYP1A2, CYP2B6, CYP2D6, CYP3A4, NR1I2, and UGT2B7: Potential Effects on Regulation by MicroRNA and Pharmacogenomics Relevance. Front. Genet. 2014, 5, 167.
- Li, M.-P.; Hu, Y.-D.; Hu, X.-L.; Zhang, Y.-J.; Yang, Y.-L.; Jiang, C.; Tang, J.; Chen, X.-P. MiRNAs and MiRNA Polymorphisms Modify Drug Response. Int. J. Environ. Res. Public Health 2016, 13, 1096.
- Zanger, U.M.; Klein, K.; Kugler, N.; Petrikat, T.; Ryu, C.S. Epigenetics and MicroRNAs in Pharmacogenetics. Adv. Pharmacol. 2018, 83, 33–64.
- Knox, B.; Wang, Y.; Rogers, L.J.; Xuan, J.; Yu, D.; Guan, H.; Chen, J.; Shi, T.; Ning, B.; Kadlubar, S.A.; et al. A Functional SNP in the 3′-UTR of TAP2 Gene Interacts with MicroRNA Hsa-miR-1270 to Suppress the Gene Expression. Environ. Mol. Mutagen. 2018, 59, 134–143.
- Yu, X.; Dhakal, I.B.; Beggs, M.; Edavana, V.K.; Williams, S.; Zhang, X.; Mercer, K.; Ning, B.; Lang, N.P.; Kadlubar, F.F.; et al. Functional Genetic Variants in the 3′-Untranslated Region of Sulfotransferase Isoform 1A1 (SULT1A1) and Their Effect on Enzymatic Activity. Toxicol. Sci. 2010, 118, 391–403.
- Omariba, G.; Xu, F.; Wang, M.; Li, K.; Zhou, Y.; Xiao, J. Genome-Wide Analysis of MicroRNA-Related Single Nucleotide Polymorphisms (SNPs) in Mouse Genome. Sci. Rep. 2020, 10, 5789.
- Zheng, Z.; Xue, F.; Wang, H.; He, Y.; Zhang, L.; Ma, W.; Zhang, C.; Guan, Y.; Ye, F.; Wen, Y.; et al. A Single Nucleotide Polymorphism-Based Formula to Predict the Risk of Propofol TCI Concentration Being over 4 µg ML−1 at the Time of Loss of Consciousness. Pharmacogenom. J. 2022, 22, 109–116.
- Clément, T.; Salone, V.; Rederstorff, M. Dual Luciferase Gene Reporter Assays to Study MiRNA Function. Methods Mol. Biol. 2015, 1296, 187–198.
- Li, S.; Xu, K.; Gu, D.; He, L.; Xie, L.; Chen, Z.; Fan, Z.; Zhu, L.; Du, M.; Chu, H.; et al. Genetic Variants in RPA1 Associated with the Response to Oxaliplatin-Based Chemotherapy in Colorectal Cancer. J. Gastroenterol. 2019, 54, 939–949.
- Hua, H.; Zeng, J.; Xing, H.; He, Y.; Han, L.; Zhang, N.; Yang, M. The RNA Editing Enzyme ADAR Modulated by the Rs1127317 Genetic Variant Diminishes EGFR-TKIs Efficiency in Advanced Lung Adenocarcinoma. Life Sci. 2022, 296, 120408.
- Papageorgiou, I.; Court, M.H. Identification and Validation of the MicroRNA Response Elements in the 3′-Untranslated Region of the UDP Glucuronosyltransferase (UGT) 2B7 and 2B15 Genes by a Functional Genomics Approach. Biochem. Pharmacol. 2017, 146, 199–213.
- Papageorgiou, I.; Court, M.H. Identification and Validation of MicroRNAs Directly Regulating the UDP-Glucuronosyltransferase 1A Subfamily Enzymes by a Functional Genomics Approach. Biochem. Pharmacol. 2017, 137, 93–106.
- Nakano, M.; Mohri, T.; Fukami, T.; Takamiya, M.; Aoki, Y.; McLeod, H.L.; Nakajima, M. Single-Nucleotide Polymorphisms in Cytochrome P450 2E1 (CYP2E1) 3′-Untranslated Region Affect the Regulation of CYP2E1 by MiR-570. Drug Metab. Dispos. 2015, 43, 1450–1457.
- Tomasello, L.; Cluts, L.; Croce, C.M. Experimental Validation of MicroRNA Targets: Mutagenesis of Binding Regions. Methods Mol. Biol. 2019, 1970, 331–339.
- Ipe, J.; Collins, K.S.; Hao, Y.; Gao, H.; Bhatia, P.; Gaedigk, A.; Liu, Y.; Skaar, T.C. PASSPORT-Seq: A Novel High-Throughput Bioassay to Functionally Test Polymorphisms in Micro-RNA Target Sites. Front Genet. 2018, 9, 219.
- Zeng, L.; Chen, Y.; Wang, Y.; Yu, L.-R.; Knox, B.; Chen, J.; Shi, T.; Chen, S.; Ren, Z.; Guo, L.; et al. MicroRNA Hsa-MiR-370-3p Suppresses the Expression and Induction of CYP2D6 by Facilitating MRNA Degradation. Biochem. Pharmacol. 2017, 140, 139–149.
- Yu, D.; Chen, S.; Li, D.; Knox, B.; Guo, L.; Ning, B. FREMSA: A Method That Provides Direct Evidence of the Interaction between MicroRNA and MRNA. Methods Mol. Biol. 2020, 2102, 557–566.
- Wei, R.; Yang, F.; Urban, T.J.; Li, L.; Chalasani, N.; Flockhart, D.A.; Liu, W. Impact of the Interaction between 3′-UTR SNPs and MicroRNA on the Expression of Human Xenobiotic Metabolism Enzyme and Transporter Genes. Front. Genet. 2012, 3, 248.
- Kheradpour, P.; Ernst, J.; Melnikov, A.; Rogov, P.; Wang, L.; Zhang, X.; Alston, J.; Mikkelsen, T.S.; Kellis, M. Systematic Dissection of Regulatory Motifs in 2000 Predicted Human Enhancers Using a Massively Parallel Reporter Assay. Genome Res. 2013, 23, 800–811.
- Li, D.; Chen, M.; Hong, H.; Tong, W.; Ning, B. Integrative Approaches for Studying the Role of Noncoding RNAs in Influencing Drug Efficacy and Toxicity. Expert. Opin. Drug Metab. Toxicol. 2022, 18, 151–163.
- Powell, N.R.; Zhao, H.; Ipe, J.; Liu, Y.; Skaar, T.C. Mapping the MiRNA-mRNA Interactome in Human Hepatocytes and Identification of Functional MirSNPs in Pharmacogenes. Clin. Pharmacol. Ther. 2021, 110, 1106–1118.
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the Human MiRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell 2013, 153, 654–665.
- Offer, S.M.; Butterfield, G.L.; Jerde, C.R.; Fossum, C.C.; Wegner, N.J.; Diasio, R.B. MicroRNAs MiR-27a and MiR-27b Directly Regulate Liver Dihydropyrimidine Dehydrogenase Expression through Two Conserved Binding Sites. Mol. Cancer Ther. 2014, 13, 742–751.


