Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Wenlu Yu | -- | 1468 | 2022-11-22 05:27:24 | | | |
| 2 | Catherine Yang | Meta information modification | 1468 | 2022-11-22 07:02:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yu, W.; Li, C.; Zhang, D.; Li, Z.; Xia, P.; Liu, X.; Cai, X.; Yang, P.; Ling, J.; Zhang, J.; et al. T Cells in Metabolic Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/35649 (accessed on 26 July 2026).
Yu W, Li C, Zhang D, Li Z, Xia P, Liu X, et al. T Cells in Metabolic Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/35649. Accessed July 26, 2026.
Yu, Wenlu, Chunxiu Li, Deju Zhang, Zhangwang Li, Panpan Xia, Xiao Liu, Xia Cai, Pingping Yang, Jitao Ling, Jing Zhang, et al. "T Cells in Metabolic Diseases" Encyclopedia, https://encyclopedia.pub/entry/35649 (accessed July 26, 2026).
Yu, W., Li, C., Zhang, D., Li, Z., Xia, P., Liu, X., Cai, X., Yang, P., Ling, J., Zhang, J., Zhang, M., & Yu, P. (2022, November 22). T Cells in Metabolic Diseases. In Encyclopedia. https://encyclopedia.pub/entry/35649
Yu, Wenlu, et al. "T Cells in Metabolic Diseases." Encyclopedia. Web. 22 November, 2022.
Copy Citation
Metabolic disease is a kind of multi-system abnormal disease which is manifested by diseases or disorders that disrupt normal metabolism, including hyperglycemia, dyslipidemia, hypertension, obesity, and insulin resistance, and leads to a dramatic increase in the occurrence of cardiovascular diseases such as acute myocardial infarction and stroke. T cells are involved in the inflammatory response, which can also regulate the development of metabolic diseases, CD4+ T cells and CD8+ T cells are mainly responsible for the role.
metabolic diseases
CD4+ T cells
CD8+ T cells
inflammation
1. CD4+ T Cells
CD4+ T cells are important in metabolic diseases. Functionally, CD4+ T cells are also known as Th cells. Under the stimulation of different antigens and cytokines, CD4+ T cells can differentiate into four subsets, including Th1, Th2, Th17, and inducible regulatory T (iTreg) cells [1][2]. Th1 cells, which secret IFN-γ, exert the function of helper cellular immunity against intracellular pathogens and viruses, and contribute to the progression of chronic inflammatory diseases [3]. Th2 cells secrete cytokines such as IL-4, IL-5, and IL-13, which promote the proliferation and differentiation of B cells, assist humoral immunity, eliminate extracellular pathogens, and play a role in allergic inflammation [4]. Th17 cells can eliminate extracellular pathogens. Similar to Th1 cells, Th17 cells promote progression of inflammation [5]. Treg cells including iTreg and nTreg inhibit inflammatory response by secreting cytokines interculin-10 (IL-10) and transforming growth factor-β (TGF-β) [4]. Generally, Th1 and Th17 cells play a critical role in promoting pro-inflammatory responses, whereas Th2 and Treg cells are highly correlated with anti-inflammatory responses [6][7] (Figure 1).
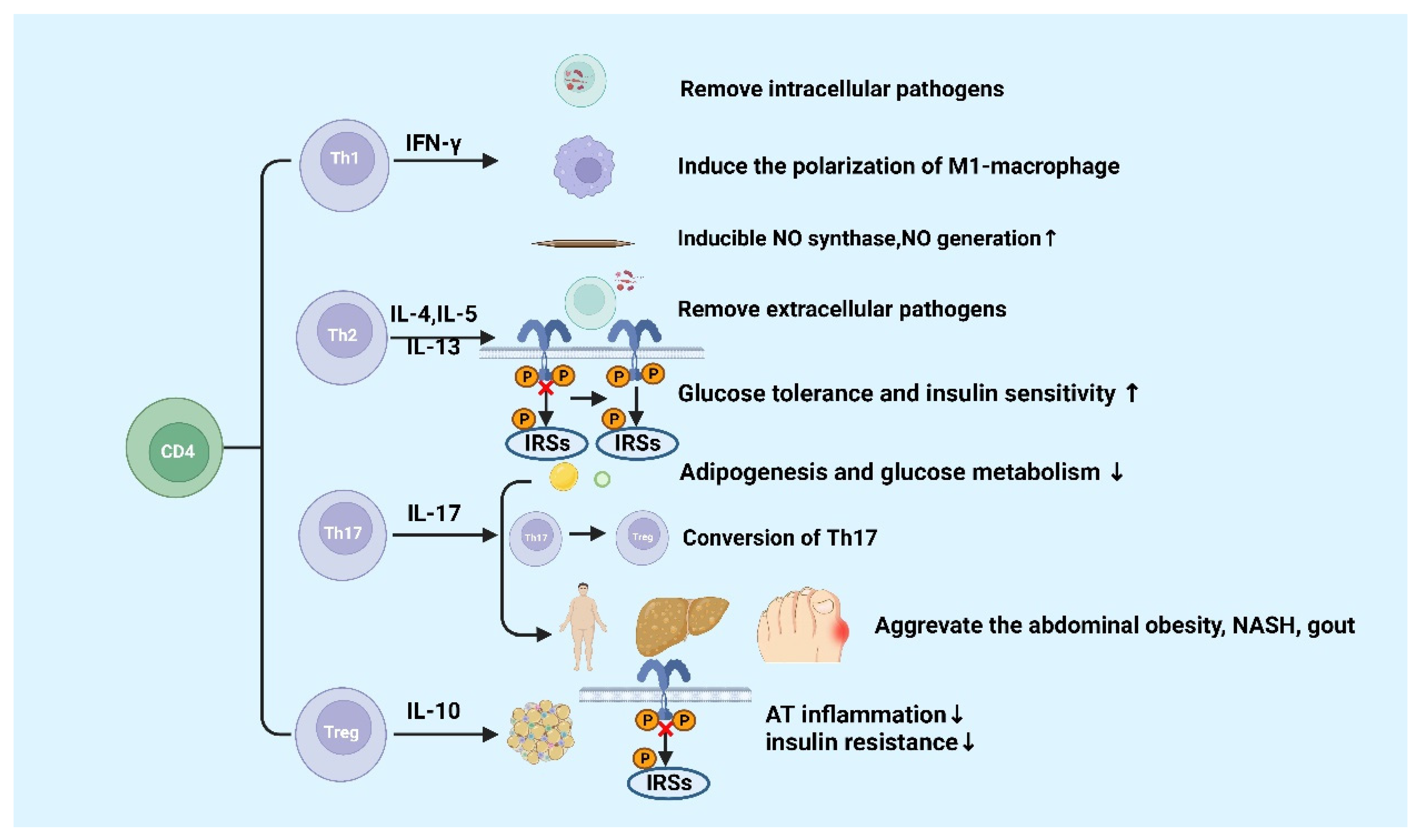
Figure 1. Functions of CD4+ T cells in metabolic diseases.
IFN-γ is produced by Th1 cells. Studies showed that the level of IFN-γ is higher in high-fat diabetes (HFD)-induced obese mice and obese populations [8]. Furthermore, IFN-γ originating from Th1 promotes the conversion of macrophages to M1 macrophages, which will exacerbate AT inflammation, promote the production of chemokines and inflammatory cytokines such as TNF-α in AT [8][9][10]. In gouty inflammation, IFN-γ could synergistically act with monosodium urate (MSU) crystals to increase the expression of inducible NO synthase and NO production in macrophages, thereby causing pro-inflammatory immune responses [11]. Ricardo-Gonzalez et al. found that in mice that suffered from allergic inflammation with Th2 bias, the glucose tolerance and insulin sensitivity greatly improved [12]. Although the function of Th-related cytokines in inflammation response in metabolic diseases has been clearly defined, the mechanisms of Th1 and Th2 cells in metabolic diseases are not well understood [13].
IL-17 is mainly secreted by Th17 cells, which suppresses glucose metabolism and adipogenesis and postpones obesity progression in mice and humans [14]. A study of Ghannam reported that IL-17 deficiency would enhance diet-induced obesity (DIO) and change glucose homeostasis. Interestingly, it was previously reported that Th17 cells can adhere to MSCs during inflammation, which contributes to their transfer to a Treg-like phenotype [15]. However, further studies found that the number of Th17 cell in abdominal subcutaneous AT in insulin-resistant individuals with obesity are higher than those insulin-sensitive individuals with obesity [16]. It was reported that inflammatory Th17 cells increased during non-alcoholic steatohepatitis (NASH) in human and mouse disease models [17][18][19]. Additionally, IL-17 expression increases cytokine production in hepatocytes [20][21]. CD4+ T cells from DIO mice could secrete high levels of IL-17. The increased levels of IL-17 induce the production of IL-8, which can regulate various immune cell infiltration into AT in response to HFD [22]. Clinical studies have discovered that level of circulating IL-17 increases in patients with gout [23][24]. These findings suggested that Th17 cells and IL-17 might be the core factor leading to metabolic diseases, except for obesity [20].
Tregs exhibit an anti-inflammatory effect via IL-10 [20], which has been confirmed in many studies. The amount of visceral adipose tissue (VAT) Treg cells significantly reduces in HFD-fed obese mice [25], showing that HFD can destroy the immune system and trigger inflammation in mice. The dramatic reduction of Treg cells in VAT has been reported to be significant in adipose inflammation and insulin resistance [25][26]. Studies have found that the increase in adipose Treg cells plays a role in attenuating adipose inflammation and promoting insulin sensitivity in HFD-fed obese mice [27][28][29]. The transfer of Treg cells reduced the expression of TNF-α and the degree of HFD-induced hepatic inflammation [30]. However, a recent study showed Tregs deficiency inhibited the progression of NASH to hepatic cell carcinoma (HCC) in a mouse model of choline deficiency, HFD feeding, and diethylnitrosamine injection [31]. Increased pro-inflammatory T cell subgroups are recorded in the natural exhaustion of Tregs in both insulin-resistant mice and T2D patients [25][32][33]. In addition, Markus Feuerer and Yaron Ilan1 have also found that inflammation and insulin resistance in mice are protected by Tregs ex vivo or in vivo [25][28]. These results indicated that Tregs suppress T2D and promote metabolic health. These results indicated that Tregs inhibit inflammation and promote insulin resistance and may participate in the progression of metabolic diseases such as obesity, NASH, and T2D further. Finally, Hyperuricemia could improve spleen Th17 proportion as well as decrease Tregs proportion in mice, thus disrupting the Th17/Tregs functional balance is crucial in gout [34]. The specific molecular mechanisms of metabolic diseases about the different roles of Th17 and Treg cells are still poorly known and require continued investigation.
Naïve CD4 T cells differentiate into different subsets in various circumstances, including Th1, Th2, Th17, and Treg cells. Generally, Th1 and Th17 cells secrete pro-inflammatory cytokines, and Th2 and Treg cells produce anti-inflammatory cytokines. The figure was constructed with BioRender (https://biorender.com, accessed on 7 September 2022).
2. CD8+ T Cells
CD8+ T cells differentiate into cytotoxic T lymphocyte (CTL) after being stimulated by antigens. CTL kills target cells by secreting cytotoxic substances such as perforin and granzyme, and induces apoptosis through the Fas/FasL pathway [32]. It was reported that CD8+ T cells play a crucial role in chronic inflammation and metabolic disorders induced by metabolic disease [35]. It has been revealed that the population of CD8+ T cells increases in the AT of both DIO and gene-induced obese mice [8][36]. Additionally, the proportion of CD4+ to CD8+ T cells decreases in obese AT prior to infiltration of macrophages [37]. Notably, the CD8+ T cells also infiltrated in human AT [35]. Moreover, in CD8-deficient obese mice, the M1 macrophages and CD8+ T cells in AT significantly reduce, AT inflammation, glucose intolerance, and the insulin resistance also decrease [32].
CD8+ T cell infiltration increase in both obese humans and mice with nonalcoholic fatty liver disease (NAFLD) [38]. In mouse models, the consumption of CD8+ T cells reduces the liver damage in NASH, indicating that CD8+ T cells may directly promote disease development [39][40]. It is reported that compared to regular NASH mice, CD8+T cell-deficient mice are associated with higher hepatic insulin sensitivity, lower liver damage, and lower fibrosis [38][39][41]. There are several probable mechanisms about activated CD8+ T cells leading to NASH development. In the early transition from steatosis to NASH, type I interferons contribute to the activation of cytotoxic CD8+ T cells, resulting in increased production of pro-inflammatory cytokines such as IFN-γ and TNF-α [41]. During the establishment of NASH, the amount of liver tissue-resistant CXCR6+CD8T cells subsets increased, which could stimulate the role of self-attack to hepatic cells and instigate the disease development [42]. Importantly, during NASH, exhausted CD8 T cells would accumulate in liver tissues, which not only damages liver tissues but impairs immunosurveillance and promotes the conversion from NASH to hepatocellular cancer [43]. However, CD8+ tissue-resident memory T cells are also beneficial for murine NASH subsiding through inducing activated hepatic stellate cells (HSCs) to apoptosis [44]. Therefore, CD8+ T cells play a potential dichotomous role in NAFLD progression and resolution.
Nishimura et al. discovered that the accumulation of CD8+ T cells results in insulin resistance and chronic inflammation in mice [32]. Chronic inflammation can increase the amount of exhausted CD8+ T cells. The amount of exhausted CD8+ T cells and the pro-inflammatory cytokines produced by them are positively correlated with fasting blood glucose. Notably, the increase in CD8+ T cells accompanied by a senile phenotype in the peripheral blood and AT may accelerate the hyperglycemia in humans [45][46]. Moreover, the research also proclaimed that the exhausted CD8+ T cells impaired insulin sensitivity in both humans and mice [45][46]. Studies showed that in DIO, chemokines secreted by CD8+ T cells that infiltrate AT induce the activation of macrophages and facilitate the recruitment of macrophages into AT [32]. Overall, these researchers prove that CD8+ T cells promote the development of metabolic diseases through pro-inflammatory activities; Figure 2 summarizes it.
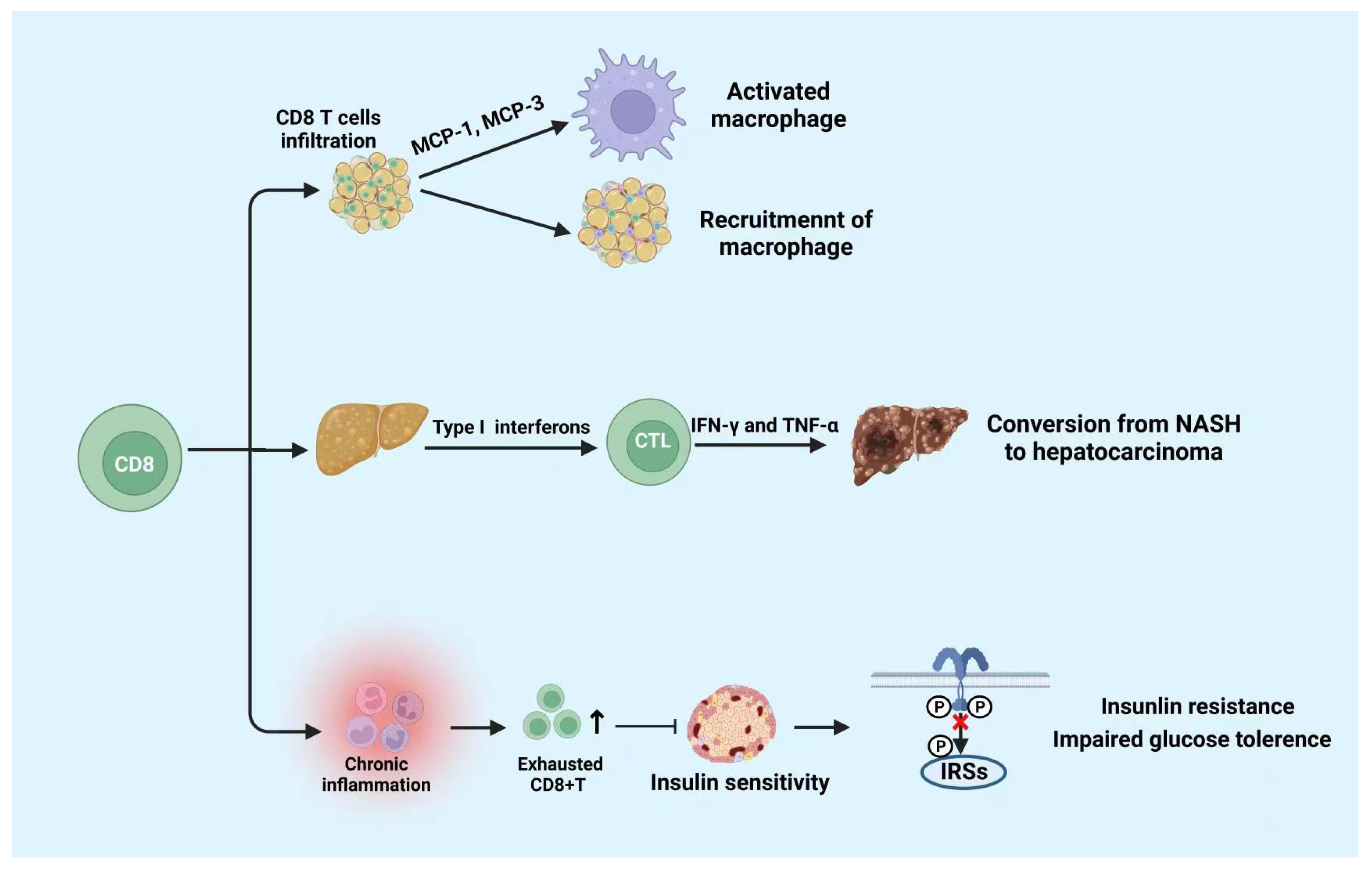
Figure 2. Functions of CD8+ T cells in metabolic diseases.
CD8+ T cells play a role in pro-inflammatory activities. CD8+ T cells infiltration promotes the activation and recruitment of macrophages. Moreover, CD8+ T cells infiltration in liver tissue is beneficial to the conversion from NASH to hepatocarcinoma. Moreover, cumulated exhausted CD8+ T cells suppress the insulin sensitivity which leads to insulin resistance and impaired glucose tolerance. NASH: non-alcoholic steatohepatitis. The figure was constructed with BioRender (https://biorender.com, accessed on 7 September 2022).
References
- O’Shea, J.J.; Paul, W.E. Mechanisms Underlying Lineage Commitment and Plasticity of Helper CD4 + T Cells. Science 2010, 327, 1098–1102.
- Reiner, S.L. Development in Motion: Helper T Cells at Work. Cell 2007, 129, 33–36.
- Hirahara, K.; Poholek, A.; Vahedi, G.; Laurence, A.; Kanno, Y.; Milner, J.D.; O’Shea, J.J. Mechanisms underlying helper T-cell plasticity: Implications for immune-mediated disease. J. Allergy Clin. Immunol. 2013, 131, 1276–1287.
- Cosmi, L.; Maggi, L.; Santarlasci, V.; Liotta, F.; Annunziato, F. T helper cells plasticity in inflammation. Cytom. Part A 2013, 85, 36–42.
- Oukka, M. Th17 cells in immunity and autoimmunity. Ann. Rheum. Dis. 2008, 67, iii26–iii29.
- Schipper, H.S.; Prakken, B.; Kalkhoven, E.; Boes, M. Adipose tissue-resident immune cells: Key players in immunometabolism. Trends Endocrinol. Metab. 2012, 23, 407–415.
- Ferrante, A.W., Jr. The immune cells in adipose tissue. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 34–38.
- Rocha, V.Z.; Folco, E.J.; Sukhova, G.; Shimizu, K.; Gotsman, I.; Vernon, A.H.; Libby, P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: A role for adaptive immunity in obesity. Circ. Res. 2008, 103, 467–476.
- Kintscher, U.; Hartge, M.; Hess, K.; Foryst-Ludwig, A.; Clemenz, M.; Wabitsch, M.; Fischer-Posovszky, P.; Barth, T.F.; Dragun, D.; Skurk, T.; et al. T-lymphocyte infiltration in visceral adipose tissue: A primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arter. Thromb. Vasc. Biol. 2008, 28, 1304–1310.
- Khan, I.M.; Perrard, X.-Y.D.; Perrard, J.L.; Mansoori, A.; Smith, C.W.; Wu, H.; Ballantyne, C.M. Attenuated adipose tissue and skeletal muscle inflammation in obese mice with combined CD4+ and CD8+ T cell deficiency. Atherosclerosis 2014, 233, 419–428.
- Jaramillo, M.; Naccache, P.H.; Olivier, M. Monosodium urate crystals synergize with IFN-gamma to generate macrophage nitric oxide: Involvement of extracellular signal-regulated kinase 1/2 and NF-kappa B. J. Immunol. 2004, 172, 5734–5742.
- Ricardo-Gonzalez, R.R.; Red Eagle, A.; Odegaard, J.I.; Jouihan, H.; Morel, C.R.; Heredia, J.E.; Mukundan, L.; Wu, D.; Locksley, R.M.; Chawla, A.; et al. IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2010, 107, 22617–22622.
- Wensveen, F.M.; Valentić, S.; Šestan, M.; Turk Wensveen, T.; Polić, B. The “Big Bang” in obese fat: Events initiating obesity-induced adipose tissue inflammation. Eur. J. Immunol. 2015, 45, 2446–2456.
- Endo, Y.; Yokote, K.; Nakayama, T. The obesity-related pathology and Th17 cells. Cell. Mol. Life Sci. 2017, 74, 1231–1245.
- Ghannam, S.; Pène, J.; Torcy-Moquet, G.; Jorgensen, C.; Yssel, H. Mesenchymal Stem Cells Inhibit Human Th17 Cell Differentiation and Function and Induce a T Regulatory Cell Phenotype. J. Immunol. 2010, 185, 302–312.
- Dalmas, E.; Venteclef, N.; Caer, C.; Poitou, C.; Cremer, I.; Aron-Wisnewsky, J.; Lacroix-Desmazes, S.; Bayry, J.; Kaveri, S.V.; Clément, K.; et al. T Cell–Derived IL-22 Amplifies IL-1β–Driven Inflammation in Human Adipose Tissue: Relevance to Obesity and Type 2 Diabetes. Diabetes 2014, 63, 1966–1977.
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92.
- Rau, M.; Schilling, A.K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105.
- Her, Z.; Tan, J.H.L.; Lim, Y.-S.; Tan, S.Y.; Chan, X.Y.; Tan, W.W.S.; Liu, M.; Yong, K.S.M.; Lai, F.; Ceccarello, E.; et al. CD4+ T Cells Mediate the Development of Liver Fibrosis in High Fat Diet-Induced NAFLD in Humanized Mice. Front. Immunol. 2020, 11, 580968.
- Zhang, S.; Gang, X.; Yang, S.; Cui, M.; Sun, L.; Li, Z.; Wang, G. The Alterations in and the Role of the Th17/Treg Balance in Metabolic Diseases. Front. Immunol. 2021, 12, 678355.
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290.
- Fossiez, F.; Djossou, O.; Chomarat, P.; Flores-Romo, L.; Ait-Yahia, S.; Maat, C.; Pin, J.J.; Garrone, P.; Garcia, E.; Saeland, S.; et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 1996, 183, 2593–2603.
- Liu, Y.; Zhao, Q.; Yin, Y.; McNutt, M.A.; Zhang, T.; Cao, Y. Serum levels of IL-17 are elevated in patients with acute gouty arthritis. Biochem. Biophys. Res. Commun. 2018, 497, 897–902.
- Yang, Q.-B.; He, Y.-L.; Zhang, Q.-B.; Mi, Q.-S.; Zhou, J.-G. Downregulation of Transcription Factor T-Bet as a Protective Strategy in Monosodium Urate-Induced Gouty Inflammation. Front. Immunol. 2019, 10, 1199.
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939.
- Deiuliis, J.; Shah, Z.; Shah, N.; Needleman, B.; Mikami, D.; Narula, V.; Perry, K.; Hazey, J.; Kampfrath, T.; Kollengode, M.; et al. Visceral Adipose Inflammation in Obesity Is Associated with Critical Alterations in Tregulatory Cell Numbers. PLoS ONE 2011, 6, e16376.
- Kolodin, D.; van Panhuys, N.; Li, C.; Magnuson, A.M.; Cipolletta, D.; Miller, C.M.; Wagers, A.; Germain, R.N.; Benoist, C.; Mathis, D. Antigen- and Cytokine-Driven Accumulation of Regulatory T Cells in Visceral Adipose Tissue of Lean Mice. Cell Metab. 2015, 21, 543–557.
- Ilan, Y.; Maron, R.; Tukpah, A.-M.; Maioli, T.U.; Murugaiyan, G.; Yang, K.; Wu, H.Y.; Weiner, H.L. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9765–9770.
- Pettersson, U.S.; Waldén, T.B.; Carlsson, P.-O.; Jansson, L.; Phillipson, M. Female Mice are Protected against High-Fat Diet Induced Metabolic Syndrome and Increase the Regulatory T Cell Population in Adipose Tissue. PLoS ONE 2012, 7, e46057.
- Ma, X.; Hua, J.; Mohamood, A.R.; Hamad, A.R.A.; Ravi, R.; Li, Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007, 46, 1519–1529.
- Wang, H.; Zhang, H.; Wang, Y.; Brown, Z.J.; Xia, Y.; Huang, Z.; Shen, C.; Hu, Z.; Beane, J.; Ansa-Addo, E.A.; et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 1271–1283.
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920.
- Jagannathan-Bogdan, M.; McDonnell, M.E.; Shin, H.; Rehman, Q.; Hasturk, H.; Apovian, C.M.; Nikolajczyk, B.S. Elevated Proinflammatory Cytokine Production by a Skewed T Cell Compartment Requires Monocytes and Promotes Inflammation in Type 2 Diabetes. J. Immunol. 2011, 186, 1162–1172.
- Liu, X.; Li, Y.; Li, Z.; Wei, X.; Ma, Y.; Cheng, P.; Jiao, R.; Fang, J.; Xing, Y.; Tang, J.; et al. A novel IgG1 monoclonal antibody against xanthine oxidase alleviates inflammation induced by potassium oxonate in mice. Int. J. Biol. Macromol. 2018, 112, 537–547.
- Wang, L.; Sun, P.; Wu, Y.; Wang, L. Metabolic tissue-resident CD8+ T cells: A key player in obesity-related diseases. Obes. Rev. 2021, 22, e13133.
- Rausch, M.E.; Weisberg, S.; Vardhana, P.; Tortoriello, D.V. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int. J. Obes. 2007, 32, 451–463.
- Anderson, E.K.; Gutierrez, D.A.; Kennedy, A.; Hasty, A.H. Weight Cycling Increases T-Cell Accumulation in Adipose Tissue and Impairs Systemic Glucose Tolerance. Diabetes 2013, 62, 3180–3188.
- Bhattacharjee, J.; Kirby, M.; Softic, S.; Miles, L.; Salazar-Gonzalez, R.-M.; Shivakumar, P.; Kohli, R. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol. Commun. 2017, 1, 299–310.
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic Activation of Intrahepatic CD8+ T Cells and NKT Cells Causes Nonalcoholic Steatohepatitis and Liver Cancer via Cross-Talk with Hepatocytes. Cancer Cell 2014, 26, 549–564.
- Van Herck, M.A.; Vonghia, L.; Kwanten, W.J.; Julé, Y.; Vanwolleghem, T.; Ebo, D.G.; Michielsen, P.P.; De Man, J.G.; Gama, L.; De Winter, B.Y.; et al. Diet Reversal and Immune Modulation Show Key Role for Liver and Adipose Tissue T Cells in Murine Nonalcoholic Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 467–490.
- Ghazarian, M.; Revelo, X.S.; Nøhr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci. Immunol. 2017, 2, eaai7616.
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Hüser, N.; Meiser, P.; Bayerl, F.; et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature 2021, 592, 444–449.
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456.
- Koda, Y.; Teratani, T.; Chu, P.-S.; Hagihara, Y.; Mikami, Y.; Harada, Y.; Tsujikawa, H.; Miyamoto, K.; Suzuki, T.; Taniki, N.; et al. CD8+ tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat. Commun. 2021, 12, 4474.
- Yi, H.-S.; Kim, S.Y.; Kim, J.T.; Lee, Y.-S.; Moon, J.S.; Kim, M.; Kang, Y.E.; Joung, K.H.; Lee, J.H.; Kim, H.J.; et al. T-cell senescence contributes to abnormal glucose homeostasis in humans and mice. Cell Death Dis. 2019, 10, 249.
- Lee, Y.-H.; Kim, S.R.; Han, D.H.; Yu, H.T.; Han, Y.D.; Kim, J.H.; Kim, S.H.; Lee, C.J.; Min, B.-H.; Kim, D.-H.; et al. Senescent T Cells Predict the Development of Hyperglycemia in Humans. Diabetes 2018, 68, 156–162.
More
Information
Subjects:
Health Policy & Services
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
22 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No