 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Long Ming | -- | 6444 | 2022-11-18 16:10:38 | | | |
| 2 | Beatrix Zheng | + 7 word(s) | 6451 | 2022-11-21 04:00:00 | | |
Video Upload Options
The mammalian target of rapamycin (mTOR) is a highly conserved serine/threonine-protein kinase, which regulates many biological processes related to metabolism, cancer, immune function, and aging. It is an essential protein kinase that belongs to the phosphoinositide-3-kinase (PI3K) family and has two known signaling complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Even though mTOR signaling plays a critical role in promoting mitochondria-related protein synthesis, suppressing the catabolic process of autophagy, contributing to lipid metabolism, engaging in ribosome formation, and acting as a critical regulator of mRNA translation, it remains one of the significant signaling systems involved in the tumor process, particularly in apoptosis, cell cycle, and cancer cell proliferation. Therefore, the mTOR signaling system could be suggested as a cancer biomarker, and its targeting is important in anti-tumor therapy research.
1. Introduction
2. mTOR and Cancer
2.1. Signalling Upstream of mTOR
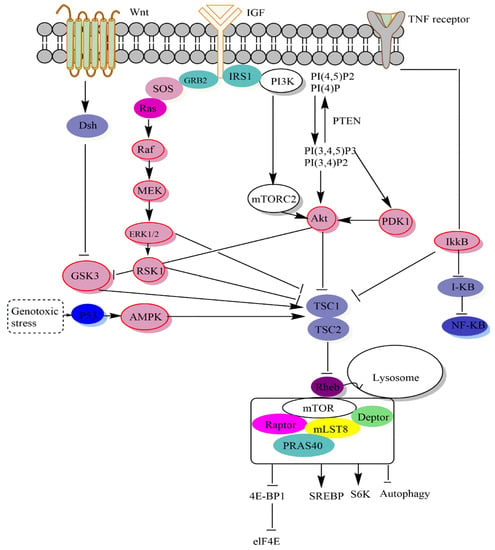
2.2. Signaling Downstream of mTOR
2.3. Breast Cancer
2.4. Lung Cancer
2.5. Colon Cancer
2.6. Head and Neck Cancer
2.7. Cervical Cancer
2.8. Reproductive Cancer
2.9. Bone Cancer
2.10. Cancers Caused by Different Type of Viruses
2.11. Nasopharyngeal Carcinoma
2.12. DLBCL
2.13. Different Type of Lymphomas
2.14. Non-Mantle Hodgkin’s Cell Lymphoma (MCL)
References
- Brown, E.J.; Albers, M.W.; Bum Shin, T.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A Mammalian Protein Targeted by G1-Arresting Rapamycin–Receptor Complex. Nature 1994, 369, 756–758.
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101.
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293.
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin Passes the Torch: A New Generation of MTOR Inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880.
- Foster, D.A. Phosphatidic Acid and Lipid-Sensing by MTOR. Trends Endocrinol. Metab. 2013, 24, 272–278.
- Huang, K.; Fingar, D.C. Growing Knowledge of the MTOR Signaling Network. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 36, pp. 79–90.
- Kim, S.M.; Vetrivel, P.; Ha, S.E.; Kim, H.H.; Kim, J.-A.; Kim, G.S. Apigetrin Induces Extrinsic Apoptosis, Autophagy and G2/M Phase Cell Cycle Arrest through PI3K/AKT/MTOR Pathway in AGS Human Gastric Cancer Cell. J. Nutr. Biochem. 2020, 83, 108427.
- Khan, K.; Quispe, C.; Javed, Z.; Iqbal, M.J.; Sadia, H.; Raza, S.; Irshad, A.; Salehi, B.; Reiner, Ž.; Sharifi-Rad, J. Resveratrol, Curcumin, Paclitaxel and MiRNAs Mediated Regulation of PI3K/Akt/MTOR Pathway: Go Four Better to Treat Bladder Cancer. Cancer Cell Int. 2020, 20, 560.
- Mafi, S.; Mansoori, B.; Taeb, S.; Sadeghi, H.; Abbasi, R.; Cho, W.C.; Rostamzadeh, D. MTOR-Mediated Regulation of Immune Responses in Cancer and Tumor Microenvironment. Front. Immunol. 2022, 12, 5724.
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory Functions of MTOR Inhibition. Nat. Rev. Immunol. 2009, 9, 324–337.
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of Innate Immune Cell Function by MTOR. Nat. Rev. Immunol. 2015, 15, 599–614.
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86.
- Weber, J. Immune Checkpoint Proteins: A New Therapeutic Paradigm for Cancer—Preclinical Background: CTLA-4 and PD-1 Blockade. In Seminars in Oncology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 37, pp. 430–439.
- Ping, W.; Liu, X.; Lei, D.; Zhang, X. MTOR Signaling-Related MicroRNAs and Cancer Involvement. J. Cancer 2018, 9, 667.
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The Coordinate Regulation of the P53 and MTOR Pathways in Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209.
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the P53 Network. Nature 2000, 408, 307–310.
- Feng, Z.; Hu, W.; De Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The Regulation of AMPK Β1, TSC2, and PTEN Expression by P53: Stress, Cell and Tissue Specificity, and the Role of These Gene Products in Modulating the IGF-1-AKT-MTOR Pathways. Cancer Res. 2007, 67, 3043–3053.
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R. Mammalian Target of Rapamycin Up-Regulation of Pyruvate Kinase Isoenzyme Type M2 Is Critical for Aerobic Glycolysis and Tumor Growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134.
- Chiang, G.G.; Abraham, R.T. Targeting the MTOR Signaling Network in Cancer. Trends Mol. Med. 2007, 13, 433–442.
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing MTOR Complexes in Regulation of Glioblastoma Multiforme and Its Stem Cells. Adv. Biol. Regul. 2013, 53, 202–210.
- Nagahashi, M.; Hait, N.C.; Maceyka, M.; Avni, D.; Takabe, K.; Milstien, S.; Spiegel, S. Sphingosine-1-Phosphate in Chronic Intestinal Inflammation and Cancer. Adv. Biol. Regul. 2014, 54, 112–120.
- Pyne, N.J.; Ohotski, J.; Bittman, R.; Pyne, S. The Role of Sphingosine 1-Phosphate in Inflammation and Cancer. Adv. Biol. Regul. 2014, 54, 121–129.
- Pyne, N.J.; Tonelli, F.; Lim, K.G.; Long, J.; Edwards, J.; Pyne, S. Targeting Sphingosine Kinase 1 in Cancer. Adv. Biol. Regul. 2012, 52, 31–38.
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 Is Phosphorylated and Inhibited by Akt and Suppresses MTOR Signalling. Nat. Cell Biol. 2002, 4, 648–657.
- Liu, L.; Li, F.; Cardelli, J.A.; Martin, K.A.; Blenis, J.; Huang, S. Rapamycin Inhibits Cell Motility by Suppression of MTOR-Mediated S6K1 and 4E-BP1 Pathways. Oncogene 2006, 25, 7029–7040.
- Vivanco, I.; Sawyers, C.L. The Phosphatidylinositol 3-Kinase–AKT Pathway in Human Cancer. Nat. Rev. Cancer 2002, 2, 489–501.
- Sansal, I.; Sellers, W.R. The Biology and Clinical Relevance of the PTEN Tumor Suppressor Pathway. J. Clin. Oncol. 2004, 22, 2954–2963.
- Stiles, B.; Groszer, M.; Wang, S.; Jiao, J.; Wu, H. PTENless Means More. Dev. Biol. 2004, 273, 175–184.
- Wymann, M.P.; Zvelebil, M.; Laffargue, M. Phosphoinositide 3-Kinase Signalling–Which Way to Target? Trends Pharmacol. Sci. 2003, 24, 366–376.
- Menon, S.; Manning, B.D. Common Corruption of the MTOR Signaling Network in Human Tumors. Oncogene 2008, 27, S43–S51.
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and Functional Inactivation of TSC2 by Erk: Implications for Tuberous Sclerosisand Cancer Pathogenesis. Cell 2005, 121, 179–193.
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt Regulates Growth by Directly Phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665.
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-Promoting Phorbol Esters and Activated Ras Inactivate the Tuberous Sclerosis Tumor Suppressor Complex via P90 Ribosomal S6 Kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494.
- Lee, D.-F.; Kuo, H.-P.; Chen, C.-T.; Hsu, J.-M.; Chou, C.-K.; Wei, Y.; Sun, H.-L.; Li, L.-Y.; Ping, B.; Huang, W.-C. IKKβ Suppression of TSC1 Links Inflammation and Tumor Angiogenesis via the MTOR Pathway. Cell 2007, 130, 440–455.
- Li, Y.; Inoki, K.; Vacratsis, P.; Guan, K.-L. The P38 and MK2 Kinase Cascade Phosphorylates Tuberin, the Tuberous Sclerosis 2 Gene Product, and Enhances Its Interaction with 14-3-3. J. Biol. Chem. 2003, 278, 13663–13671.
- Tee, A.R.; Anjum, R.; Blenis, J. Inactivation of the Tuberous Sclerosis Complex-1 and-2 Gene Products Occurs by Phosphoinositide 3-Kinase/Akt-Dependent and-Independent Phosphorylation of Tuberin. J. Biol. Chem. 2003, 278, 37288–37296.
- Dalle Pezze, P.; Ruf, S.; Sonntag, A.G.; Langelaar-Makkinje, M.; Hall, P.; Heberle, A.M.; Razquin Navas, P.; Van Eunen, K.; Tölle, R.C.; Schwarz, J.J. A Systems Study Reveals Concurrent Activation of AMPK and MTOR by Amino Acids. Nat. Commun. 2016, 7, 1–19.
- Efeyan, A.; Zoncu, R.; Chang, S.; Gumper, I.; Snitkin, H.; Wolfson, R.L.; Kirak, O.; Sabatini, D.D.; Sabatini, D.M. Regulation of MTORC1 by the Rag GTPases Is Necessary for Neonatal Autophagy and Survival. Nature 2013, 493, 679–683.
- Ghislat, G.; Patron, M.; Rizzuto, R.; Knecht, E. Withdrawal of Essential Amino Acids Increases Autophagy by a Pathway Involving Ca2+/Calmodulin-Dependent Kinase Kinase-β (CaMKK-β). J. Biol. Chem. 2012, 287, 38625–38636.
- Andjelkovic, M.; Alessi, D.R.; Meier, R.; Fernandez, A.; Lamb, N.J.; Frech, M.; Cron, P.; Cohen, P.; Lucocq, J.M.; Hemmings, B.A. Role of Translocation in the Activation and Function of Protein Kinase B. J. Biol. Chem. 1997, 272, 31515–31524.
- Frech, M.; Andjelkovic, M.; Ingley, E.; Reddy, K.K.; Falck, J.R.; Hemmings, B.A. High Affinity Binding of Inositol Phosphates and Phosphoinositides to the Pleckstrin Homology Domain of RAC/Protein Kinase B and Their Influence on Kinase Activity. J. Biol. Chem. 1997, 272, 8474–8481.
- Ma, X.M.; Blenis, J. Molecular Mechanisms of MTOR-Mediated Translational Control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318.
- Brunn, G.J.; Hudson, C.C.; Sekulic, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Lawrence, J.C., Jr.; Abraham, R.T. Phosphorylation of the Translational Repressor PHAS-I by the Mammalian Target of Rapamycin. Science 1997, 277, 99–101.
- Gingras, A.-C.; Raught, B.; Sonenberg, N. Regulation of Translation Initiation by FRAP/MTOR. Genes Dev. 2001, 15, 807–826.
- Hara, K.; Yonezawa, K.; Kozlowski, M.T.; Sugimoto, T.; Andrabi, K.; Weng, Q.-P.; Kasuga, M.; Nishimoto, I.; Avruch, J. Regulation of EIF-4E BP1 Phosphorylation by MTOR. J. Biol. Chem. 1997, 272, 26457–26463.
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP Activity Is Regulated by MTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab. 2008, 8, 224–236.
- Castilho, R.M.; Squarize, C.H.; Chodosh, L.A.; Williams, B.O.; Gutkind, J.S. MTOR Mediates Wnt-Induced Epidermal Stem Cell Exhaustion and Aging. Cell Stem Cell 2009, 5, 279–289.
- Laplante, M.; Sabatini, D.M. An Emerging Role of MTOR in Lipid Biosynthesis. Curr. Biol. 2009, 19, R1046–R1052.
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. MTOR: From Growth Signal Integration to Cancer, Diabetes and Ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35.
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 Phosphorylation of the Translational Regulators P70 S6 Kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437.
- Brown, E.J.; Beal, P.A.; Keith, C.T.; Chen, J.; Bum Shin, T.; Schreiber, S.L. Control of P70 S6 Kinase by Kinase Activity of FRAP in Vivo. Nature 1995, 377, 441–446.
- Gingras, A.-C.; Raught, B.; Sonenberg, N. EIF4 Initiation Factors: Effectors of MRNA Recruitment to Ribosomes and Regulators of Translation. Annu. Rev. Biochem. 1999, 68, 913–963.
- Hay, N.; Sonenberg, N. Upstream and Downstream of MTOR. Genes Dev. 2004, 18, 1926–1945.
- Wang, X.; Beugnet, A.; Murakami, M.; Yamanaka, S.; Proud, C.G. Distinct Signaling Events Downstream of MTOR Cooperate to Mediate the Effects of Amino Acids and Insulin on Initiation Factor 4E-Binding Proteins. Mol. Cell Biol. 2005, 25, 2558–2572.
- Yellen, P.; Saqcena, M.; Salloum, D.; Feng, J.; Preda, A.; Xu, L.; Rodrik-Outmezguine, V.; Foster, D.A. High-Dose Rapamycin Induces Apoptosis in Human Cancer Cells by Dissociating MTOR Complex 1 and Suppressing Phosphorylation of 4E-BP1. Cell Cycle 2011, 10, 3948–3956.
- Bärlund, M.; Monni, O.; Kononen, J.; Cornelison, R.; Torhorst, J.; Sauter, G.; Kallioniemi, O.-P.; Kallioniemi, A. Multiple Genes at 17q23 Undergo Amplification and Overexpression in Breast Cancer. Cancer Res. 2000, 60, 5340–5344.
- Surace, E.I.; Lusis, E.; Haipek, C.A.; Gutmann, D.H. Functional Significance of S6K Overexpression in Meningioma Progression. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2004, 56, 295–298.
- Kim, J.E.; Chen, J. Regulation of Peroxisome Proliferator–Activated Receptor-γ Activity by Mammalian Target of Rapamycin and Amino Acids in Adipogenesis. Diabetes 2004, 53, 2748–2756.
- Armour, S.M.; Baur, J.A.; Hsieh, S.N.; Land-Bracha, A.; Thomas, S.M.; Sinclair, D.A. Inhibition of Mammalian S6 Kinase by Resveratrol Suppresses Autophagy. Aging 2009, 1, 515.
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.D.; Blenis, J. Mammalian Cell Size Is Controlled by MTOR and Its Downstream Targets S6K1 and 4EBP1/EIF4E. Genes Dev. 2002, 16, 1472–1487.
- Wang, J.; Sheng, Z.; Cai, Y. Effects of MicroRNA-513b on Cell Proliferation, Apoptosis, Invasion, and Migration by Targeting HMGB3 through Regulation of MTOR Signaling Pathway in Non-Small-Cell Lung Cancer. J. Cell Physiol. 2019, 234, 10934–10941.
- Sun, J.; Chen, Z.; Tan, X.; Zhou, F.; Tan, F.; Gao, Y.; Sun, N.; Xu, X.; Shao, K.; He, J. MicroRNA-99a/100 Promotes Apoptosis by Targeting MTOR in Human Esophageal Squamous Cell Carcinoma. Med. Oncol. 2013, 30, 1–9.
- Liao, W.; Zhang, Y. MicroRNA-381 Facilitates Autophagy and Apoptosis in Prostate Cancer Cells via Inhibiting the RELN-Mediated PI3K/AKT/MTOR Signaling Pathway. Life Sci. 2020, 254, 117672.
- Liu, T.; Ye, P.; Ye, Y.; Han, B. MicroRNA-216b Targets HK2 to Potentiate Autophagy and Apoptosis of Breast Cancer Cells via the MTOR Signaling Pathway. Int. J. Biol. Sci. 2021, 17, 2970.
- Li, X.J.; Luo, X.Q.; Han, B.W.; Duan, F.T.; Wei, P.P.; Chen, Y.Q. MicroRNA-100/99a, Deregulated in Acute Lymphoblastic Leukaemia, Suppress Proliferation and Promote Apoptosis by Regulating the FKBP51 and IGF1R/MTOR Signalling Pathways. Br. J. Cancer 2013, 109, 2189–2198.
- Ueng, S.-H.; Chen, S.-C.; Chang, Y.-S.; Hsueh, S.; Lin, Y.-C.; Chien, H.-P.; Lo, Y.-F.; Shen, S.-C.; Hsueh, C. Phosphorylated MTOR Expression Correlates with Poor Outcome in Early-Stage Triple Negative Breast Carcinomas. Int. J. Clin. Exp. Pathol. 2012, 5, 806.
- Xu, K.; Liu, P.; Wei, W. MTOR Signaling in Tumorigenesis. Biochim. Biophys. Acta BBA-Rev. Cancer 2014, 1846, 638–654.
- Krieger, K.L.; Hu, W.-F.; Ripperger, T.; Woods, N.T. Functional Impacts of the BRCA1-MTORC2 Interaction in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 5876.
- Woods, N.T.; Mesquita, R.D.; Sweet, M.; Carvalho, M.A.; Li, X.; Liu, Y.; Nguyen, H.; Thomas, C.E.; Iversen, E.S., Jr.; Marsillac, S. Charting the Landscape of Tandem BRCT Domain–Mediated Protein Interactions. Sci. Signal. 2012, 5, rs6.
- Wazir, U.; Newbold, R.F.; Jiang, W.G.; Sharma, A.K.; Mokbel, K. Prognostic and Therapeutic Implications of MTORC1 and Rictor Expression in Human Breast Cancer. Oncol. Rep. 2013, 29, 1969–1974.
- Dogan, F.; Avci, C.B. Correlation between Telomerase and MTOR Pathway in Cancer Stem Cells. Gene 2018, 641, 235–239.
- Hynes, N.E.; Boulay, A. The MTOR Pathway in Breast Cancer. J. Mammary Gland Biol. Neoplasia 2006, 11, 53–61.
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C. Mutations and Deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/MTOR Cascades Which Alter Therapy Response. Oncotarget 2012, 3, 954.
- Strimpakos, A.S.; Karapanagiotou, E.M.; Saif, M.W.; Syrigos, K.N. The Role of MTOR in the Management of Solid Tumors: An Overview. Cancer Treat. Rev. 2009, 35, 148–159.
- Walsh, S.; Flanagan, L.; Quinn, C.; Evoy, D.; McDermott, E.W.; Pierce, A.; Duffy, M.J. MTOR in Breast Cancer: Differential Expression in Triple-Negative and Non-Triple-Negative Tumors. Breast 2012, 21, 178–182.
- Droog, M.; Beelen, K.; Linn, S.; Zwart, W. Tamoxifen Resistance: From Bench to Bedside. Eur. J. Pharmacol. 2013, 717, 47–57.
- Viedma-Rodríguez, R.; Baiza-Gutman, L.; Salamanca-Gómez, F.; Diaz-Zaragoza, M.; Martínez-Hernández, G.; Ruiz Esparza-Garrido, R.; Velázquez-Flores, M.A.; Arenas-Aranda, D. Mechanisms Associated with Resistance to Tamoxifen in Estrogen Receptor-Positive Breast Cancer. Oncol. Rep. 2014, 32, 3–15.
- Hare, S.H.; Harvey, A.J. MTOR Function and Therapeutic Targeting in Breast Cancer. Am. J. Cancer Res. 2017, 7, 383.
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 1–9.
- Brady, S.W.; Zhang, J.; Tsai, M.-H.; Yu, D. PI3K-Independent MTOR Activation Promotes Lapatinib Resistance and IAP Expression That Can Be Effectively Reversed by MTOR and Hsp90 Inhibition. Cancer Biol. Ther. 2015, 16, 402–411.
- Margariti, N.; Fox, S.B.; Bottini, A.; Generali, D. “Overcoming Breast Cancer Drug Resistance with MTOR Inhibitors”. Could It Be a Myth or a Real Possibility in the Short-Term Future? Breast Cancer Res. Treat. 2011, 128, 599–606.
- Liu, Y.; Mao, C.; Wang, M.; Liu, N.; Ouyang, L.; Liu, S.; Tang, H.; Cao, Y.; Liu, S.; Wang, X. Cancer Progression Is Mediated by Proline Catabolism in Non-Small Cell Lung Cancer. Oncogene 2020, 39, 2358–2376.
- Schettino, C.; Bareschino, M.A.; Sacco, P.C.; Maione, P.; Rossi, A.; Casaluce, F.; Sgambato, A.; Gridelli, C. New Molecular Targets in the Treatment of NSCLC. Curr. Pharm. Des. 2013, 19, 5333–5343.
- Pothongsrisit, S.; Pongrakhananon, V. Targeting the PI3K/AKT/MTOR Signaling Pathway in Lung Cancer: An Update Regarding Potential Drugs and Natural Products. Molecules 2021, 26, 4100.
- Ekman, S.; Wynes, M.W.; Hirsch, F.R. The MTOR Pathway in Lung Cancer and Implications for Therapy and Biomarker Analysis. J. Thorac. Oncol. 2012, 7, 947–953.
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B. Somatic Mutations Affect Key Pathways in Lung Adenocarcinoma. Nature 2008, 455, 1069–1075.
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the Phosphoinositide 3-Kinase Pathway in Cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644.
- Zhou, W.; Marcus, A.I.; Vertino, P.M. Dysregulation of MTOR Activity through LKB1 Inactivation. Chin. J. Cancer 2013, 32, 427.
- Mamane, Y.; Petroulakis, E.; Rong, L.; Yoshida, K.; Ler, L.W.; Sonenberg, N. EIF4E–from Translation to Transformation. Oncogene 2004, 23, 3172–3179.
- Rosenwald, I.B.; Hutzler, M.J.; Wang, S.; Savas, L.; Fraire, A.E. Expression of Eukaryotic Translation Initiation Factors 4E and 2α Is Increased Frequently in Bronchioloalveolar but Not in Squamous Cell Carcinomas of the Lung. Cancer 2001, 92, 2164–2171.
- Seki, N.; Takasu, T.; Mandai, K.; Nakata, M.; Saeki, H.; Heike, Y.; Takata, I.; Segawa, Y.; Hanafusa, T.; Eguchi, K. Expression of Eukaryotic Initiation Factor 4E in Atypical Adenomatous Hyperplasia and Adenocarcinoma of the Human Peripheral Lung. Clin. Cancer Res. 2002, 8, 3046–3053.
- Frankel, S.K.; Moats-Staats, B.M.; Cool, C.D.; Wynes, M.W.; Stiles, A.D.; Riches, D.W. Human Insulin-like Growth Factor-IA Expression in Transgenic Mice Promotes Adenomatous Hyperplasia but Not Pulmonary Fibrosis. Am. J. Physiol.-Lung Cell Mol. Physiol. 2005, 288, L805–L812.
- Conde, E.; Angulo, B.; Tang, M.; Morente, M.; Torres-Lanzas, J.; Lopez-Encuentra, A.; Lopez-Rios, F.; Sanchez-Cespedes, M. Molecular Context of the EGFR Mutations: Evidence for the Activation of MTOR/S6K Signaling. Clin. Cancer Res. 2006, 12, 710–717.
- Faoro, L.; Singleton, P.A.; Cervantes, G.M.; Lennon, F.E.; Choong, N.W.; Kanteti, R.; Ferguson, B.D.; Husain, A.N.; Tretiakova, M.S.; Ramnath, N. EphA2 Mutation in Lung Squamous Cell Carcinoma Promotes Increased Cell Survival, Cell Invasion, Focal Adhesions, and Mammalian Target of Rapamycin Activation. J. Biol. Chem. 2010, 285, 18575–18585.
- Dobashi, Y.; Suzuki, S.; Matsubara, H.; Kimura, M.; Endo, S.; Ooi, A. Critical and Diverse Involvement of Akt/Mammalian Target of Rapamycin Signaling in Human Lung Carcinomas. Cancer 2009, 115, 107–118.
- Wang, H.; Liu, Y.; Ding, J.; Huang, Y.; Liu, J.; Liu, N.; Ao, Y.; Hong, Y.; Wang, L.; Zhang, L. Targeting MTOR Suppressed Colon Cancer Growth through 4EBP1/EIF4E/PUMA Pathway. Cancer Gene Ther. 2020, 27, 448–460.
- Francipane, M.G.; Lagasse, E. MTOR Pathway in Colorectal Cancer: An Update. Oncotarget 2014, 5, 49.
- Gulhati, P.; Cai, Q.; Li, J.; Liu, J.; Rychahou, P.G.; Qiu, S.; Lee, E.Y.; Silva, S.R.; Bowen, K.A.; Gao, T. Targeted Inhibition of Mammalian Target of Rapamycin Signaling Inhibits Tumorigenesis of Colorectal CancerTargeting MTOR Signaling in Colorectal Cancer. Clin. Cancer Res. 2009, 15, 7207–7216.
- He, K.; Zheng, X.; Li, M.; Zhang, L.; Yu, J. MTOR Inhibitors Induce Apoptosis in Colon Cancer Cells via CHOP-Dependent DR5 Induction on 4E-BP1 Dephosphorylation. Oncogene 2016, 35, 148–157.
- Zhang, Y.-J.; Dai, Q.; Sun, D.-F.; Xiong, H.; Tian, X.-Q.; Gao, F.-H.; Xu, M.-H.; Chen, G.-Q.; Han, Z.-G.; Fang, J.-Y. MTOR Signaling Pathway Is a Target for the Treatment of Colorectal Cancer. Ann. Surg. Oncol. 2009, 16, 2617–2628.
- Roulin, D.; Cerantola, Y.; Dormond-Meuwly, A.; Demartines, N.; Dormond, O. Targeting MTORC2 Inhibits Colon Cancer Cell Proliferation in Vitro and Tumor Formation in Vivo. Mol. Cancer 2010, 9, 1–4.
- Gulhati, P.; Bowen, K.A.; Liu, J.; Stevens, P.D.; Rychahou, P.G.; Chen, M.; Lee, E.Y.; Weiss, H.L.; O’Connor, K.L.; Gao, T. MTORC1 and MTORC2 Regulate EMT, Motility, and Metastasis of Colorectal Cancer via RhoA and Rac1 Signaling PathwaysmTOR Signaling in EMT and Metastasis of CRC. Cancer Res. 2011, 71, 3246–3256.
- Aoki, K.; Tamai, Y.; Horiike, S.; Oshima, M.; Taketo, M.M. Colonic Polyposis Caused by MTOR-Mediated Chromosomal Instability in Apc+/Δ716 Cdx2+/- Compound Mutant Mice. Nat. Genet. 2003, 35, 323–330.
- Wang, D.; Xu, C.; Yang, W.; Chen, J.; Ou, Y.; Guan, Y.; Guan, J.; Liu, Y. E3 Ligase RNF167 and Deubiquitinase STAMBPL1 Modulate MTOR and Cancer Progression. Mol. Cell 2022, 82, 770–784.
- Weng, M.; Chen, W.; Chen, X.; Lu, H.; Sun, Z.; Yu, Q.; Sun, P.; Xu, Y.; Zhu, M.; Jiang, N. Fasting Inhibits Aerobic Glycolysis and Proliferation in Colorectal Cancer via the Fdft1-Mediated AKT/MTOR/HIF1α Pathway Suppression. Nat. Commun. 2020, 11, 1–17.
- Wang, K.; Huang, W.; Sang, X.; Wu, X.; Shan, Q.; Tang, D.; Xu, X.; Cao, G. Atractylenolide I Inhibits Colorectal Cancer Cell Proliferation by Affecting Metabolism and Stemness via AKT/MTOR Signaling. Phytomedicine 2020, 68, 153191.
- Roper, J.; Richardson, M.P.; Wang, W.V.; Richard, L.G.; Chen, W.; Coffee, E.M.; Sinnamon, M.J.; Lee, L.; Chen, P.-C.; Bronson, R.T. The Dual PI3K/MTOR Inhibitor NVP-BEZ235 Induces Tumor Regression in a Genetically Engineered Mouse Model of PIK3CA Wild-Type Colorectal Cancer. PLoS ONE 2011, 6, e25132.
- Belmont, P.J.; Jiang, P.; McKee, T.D.; Xie, T.; Isaacson, J.; Baryla, N.E.; Roper, J.; Sinnamon, M.J.; Lee, N.V.; Kan, J.L. Resistance to Dual Blockade of the Kinases PI3K and MTOR in KRAS-Mutant Colorectal Cancer Models Results in Combined Sensitivity to Inhibition of the Receptor Tyrosine Kinase EGFR. Sci. Signal. 2014, 7, ra107.
- Shen, J.; Wang, A.; Wang, Q.; Gurvich, I.; Siegel, A.B.; Remotti, H.; Santella, R.M. Exploration of Genome-Wide Circulating MicroRNA in Hepatocellular Carcinoma: MiR-483-5p as a Potential Biomarker. Cancer Epidemiol. Prev. Biomark. 2013, 22, 2364–2373.
- Tan, F.H.; Bai, Y.; Saintigny, P.; Darido, C. MTOR Signalling in Head and Neck Cancer: Heads Up. Cells 2019, 8, 333.
- Tian, T.; Li, X.; Zhang, J. MTOR Signaling in Cancer and MTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755.
- Sun, Z.-J.; Zhang, L.; Hall, B.; Bian, Y.; Gutkind, J.S.; Kulkarni, A.B. Chemopreventive and Chemotherapeutic Actions of MTOR Inhibitor in Genetically Defined Head and Neck Squamous Cell Carcinoma Mouse ModelRapamycin Prevents Tumorigenesis in HNSCC Mouse Model. Clin. Cancer Res. 2012, 18, 5304–5313.
- Moore, E.C.; Cash, H.A.; Caruso, A.M.; Uppaluri, R.; Hodge, J.W.; Van Waes, C.; Allen, C.T. Enhanced Tumor Control with Combination MTOR and PD-L1 Inhibition in Syngeneic Oral Cavity CancersCombination MTOR and PD-L1 Checkpoint Inhibition. Cancer Immunol. Res. 2016, 4, 611–620.
- Bozec, A.; Ebran, N.; Radosevic-Robin, N.; Sudaka, A.; Monteverde, M.; Toussan, N.; Etienne-Grimaldi, M.-C.; Nigro, C.L.; Merlano, M.; Penault-Llorca, F. Combination of m TOR and EGFR Targeting in an Orthotopic Xenograft Model of Head and Neck Cancer. Laryngoscope 2016, 126, E156–E163.
- Li, Z.; Yang, Z.; Passaniti, A.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H.C. A Positive Feedback Loop Involving EGFR/Akt/MTORC1 and IKK/NF-ΚB Regulates Head and Neck Squamous Cell Carcinoma Proliferation. Oncotarget 2016, 7, 31892.
- Suda, T.; Hama, T.; Kondo, S.; Yuza, Y.; Yoshikawa, M.; Urashima, M.; Kato, T.; Moriyama, H. Copy Number Amplification of the PIK3CA Gene Is Associated with Poor Prognosis in Non-Lymph Node Metastatic Head and Neck Squamous Cell Carcinoma. BMC Cancer 2012, 12, 1–9.
- Keysar, S.B.; Le, P.N.; Miller, B.; Jackson, B.C.; Eagles, J.R.; Nieto, C.; Kim, J.; Tang, B.; Glogowska, M.J.; Morton, J.J. Regulation of Head and Neck Squamous Cancer Stem Cells by PI3K and SOX2. J. Natl. Cancer Inst. 2017, 109, djw189.
- Pedrero, J.M.G.; Carracedo, D.G.; Pinto, C.M.; Zapatero, A.H.; Rodrigo, J.P.; Nieto, C.S.; Gonzalez, M.V. Retracted: Frequent Genetic and Biochemical Alterations of the PI 3-K/AKT/PTEN Pathway in Head and Neck Squamous Cell Carcinoma. Int. J. Cancer 2005, 114, 242–248.
- Georgy, S.R.; Cangkrama, M.; Srivastava, S.; Partridge, D.; Auden, A.; Dworkin, S.; McLean, C.A.; Jane, S.M.; Darido, C. Identification of a Novel Proto-Oncogenic Network in Head and Neck Squamous Cell Carcinoma. JNCI J. Natl. Cancer Inst. 2015, 107.
- Bian, Y.; Hall, B.; Sun, Z.-J.; Molinolo, A.; Chen, W.; Gutkind, J.S.; Waes, C.V.; Kulkarni, A.B. Loss of TGF-β Signaling and PTEN Promotes Head and Neck Squamous Cell Carcinoma through Cellular Senescence Evasion and Cancer-Related Inflammation. Oncogene 2012, 31, 3322–3332.
- Kiaris, H.; Spandidos, D.A.; Jones, A.S.; Vaughan, E.D.; Field, J.K. Mutations, Expression and Genomic Instability of the H-Ras Proto-Oncogene in Squamous Cell Carcinomas of the Head and Neck. Br. J. Cancer 1995, 72, 123–128.
- Ruicci, K.M.; Pinto, N.; Khan, M.I.; Yoo, J.; Fung, K.; MacNeil, D.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C. ERK-TSC2 Signalling in Constitutively-Active HRAS Mutant HNSCC Cells Promotes Resistance to PI3K Inhibition. Oral Oncol. 2018, 84, 95–103.
- Feng, W.; Duan, X.; Liu, J.; Xiao, J.; Brown, R.E. Morphoproteomic Evidence of Constitutively Activated and Overexpressed MTOR Pathway in Cervical Squamous Carcinoma and High Grade Squamous Intraepithelial Lesions. Int. J. Clin. Exp. Pathol. 2009, 2, 249.
- Harada, H.; Andersen, J.S.; Mann, M.; Terada, N.; Korsmeyer, S.J. P70S6 Kinase Signals Cell Survival as Well as Growth, Inactivating the pro-Apoptotic Molecule BAD. Proc. Natl. Acad. Sci. USA 2001, 98, 9666–9670.
- Ji, J.; Zheng, P.-S. Activation of MTOR Signaling Pathway Contributes to Survival of Cervical Cancer Cells. Gynecol. Oncol. 2010, 117, 103–108.
- Faried, L.S.; Faried, A.; Kanuma, T.; Sano, T.; Nakazato, T.; Tamura, T.; Kuwano, H.; Minegishi, T. Predictive and Prognostic Role of Activated Mammalian Target of Rapamycin in Cervical Cancer Treated with Cisplatin-Based Neoadjuvant Chemotherapy. Oncol. Rep. 2006, 16, 57–63.
- Wang, F.; Tan, W.H.; Liu, W.; Jin, Y.X.; Dong, D.D.; Zhao, X.J.; Liu, Q. Effects of MiR-214 on Cervical Cancer Cell Proliferation, Apoptosis and Invasion via Modulating PI3K/AKT/MTOR Signal Pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1891–1898.
- Liu, L.; Wang, Y.; Geng, C.; Wang, A.; Han, S.; You, X.; Sun, Y.; Zhang, J.; Lu, W.; Zhang, Y. CD155 Promotes the Progression of Cervical Cancer Cells Through AKT/MTOR and NF-ΚB Pathways. Front. Oncol. 2021, 2153.
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/MTOR Signaling Pathway Alleviates Ovarian Cancer Chemoresistance through Reversing Epithelial-Mesenchymal Transition and Decreasing Cancer Stem Cell Marker Expression. BMC Cancer 2019, 19, 1–12.
- Bi, X.; Lv, X.; Liu, D.; Guo, H.; Yao, G.; Wang, L.; Liang, X.; Yang, Y. METTL3-Mediated Maturation of MiR-126-5p Promotes Ovarian Cancer Progression via PTEN-Mediated PI3K/Akt/MTOR Pathway. Cancer Gene Ther. 2021, 28, 335–349.
- Zhou, Z.; Tu, Z.; Zhang, J.; Shen, X.; Wan, B.; Li, Y.; Wang, A.; Zhao, L.; Hu, J.; Ma, N. Follicular Fluid-Derived Exosomal MicroRNA-18b-5p Regulates PTEN-Mediated PI3K/Akt/MTOR Signaling Pathway to Inhibit Polycystic Ovary Syndrome Development. Mol. Neurobiol. 2022, 59, 2520–2531.
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Acquisition of Epithelial–Mesenchymal Transition and Cancer Stem Cell Phenotypes Is Associated with Activation of the PI3K/Akt/MTOR Pathway in Prostate Cancer Radioresistance. Cell Death Dis. 2013, 4, e875.
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-MTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507.
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers 2017, 9, 34.
- Malik, S.N.; Brattain, M.; Ghosh, P.M.; Troyer, D.A.; Prihoda, T.; Bedolla, R.; Kreisberg, J.I. Immunohistochemical Demonstration of Phospho-Akt in High Gleason Grade Prostate Cancer. Clin. Cancer Res. 2002, 8, 1168–1171.
- Bertoldo, F.; Silvestris, F.; Ibrahim, T.; Cognetti, F.; Generali, D.; Ripamonti, C.I.; Amadori, D.; Colleoni, M.A.; Conte, P.; Del Mastro, L. Targeting Bone Metastatic Cancer: Role of the MTOR Pathway. Biochim. Biophys. Acta BBA—Rev. Cancer 2014, 1845, 248–254.
- Zhang, J.; Wang, L.; Wang, H.; Su, Z.; Pang, X. Neuroinflammation and Central PI3K/Akt/MTOR Signal Pathway Contribute to Bone Cancer Pain. Mol. Pain 2019, 15, 1744806919830240.
- Ma, J.; Li, M.; Hock, J.; Yu, X. Hyperactivation of MTOR Critically Regulates Abnormal Osteoclastogenesis in Neurofibromatosis Type 1. J. Orthop. Res. 2012, 30, 144–152.
- Ding, L.; Congwei, L.; Bei, Q.; Tao, Y.; Ruiguo, W.; Heze, Y.; Bo, D.; Zhihong, L. MTOR: An Attractive Therapeutic Target for Osteosarcoma? Oncotarget 2016, 7, 50805.
- Martin, D.; Gutkind, J.S. Human Tumor-Associated Viruses and New Insights into the Molecular Mechanisms of Cancer. Oncogene 2008, 27, S31–S42.
- Moore, P.S.; Chang, Y. Why Do Viruses Cause Cancer? Highlights of the First Century of Human Tumour Virology. Nat. Rev. Cancer 2010, 10, 878–889.
- Zhang, L.; Wu, J.; Ling, M.T.; Zhao, L.; Zhao, K.-N. The Role of the PI3K/Akt/MTOR Signalling Pathway in Human Cancers Induced by Infection with Human Papillomaviruses. Mol. Cancer 2015, 14, 1–13.
- Sewell, A.; Brown, B.; Biktasova, A.; Mills, G.B.; Lu, Y.; Tyson, D.R.; Issaeva, N.; Yarbrough, W.G. Reverse-Phase Protein Array Profiling of Oropharyngeal Cancer and Significance of PIK3CA Mutations in HPV-Associated Head and Neck CancerMutant PIK3CA in Head and Neck Cancer. Clin. Cancer Res. 2014, 20, 2300–2311.
- Surviladze, Z.; Sterk, R.T.; DeHaro, S.A.; Ozbun, M.A. Cellular Entry of Human Papillomavirus Type 16 Involves Activation of the Phosphatidylinositol 3-Kinase/Akt/MTOR Pathway and Inhibition of Autophagy. J. Virol. 2013, 87, 2508–2517.
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C Virus Upregulates Beclin1 for Induction of Autophagy and Activates MTOR Signaling. J. Virol. 2012, 86, 8705–8712.
- Aravinth, S.P.; Rajendran, S.; Li, Y.; Wu, M.; Wong, A.H.Y.; Schwarz, H. Epstein–Barr Virus-Encoded LMP1 Induces Ectopic CD137 Expression on Hodgkin and Reed–Sternberg Cells via the PI3K-AKT-MTOR Pathway. Leuk. Lymphoma 2019, 60, 2697–2704.
- Wang, W.; Wen, Q.; Xu, L.; Xie, G.; Li, J.; Luo, J.; Chu, S.; Shi, L.; Huang, D.; Li, J. Activation of Akt/MTOR Pathway Is Associated with Poor Prognosis of Nasopharyngeal Carcinoma. PLoS ONE 2014, 9, e106098.
- Wang, Y.; Sun, J.; Yao, N. Correlation of the AKT/MTOR Signaling Pathway with the Clinicopathological Features and Prognosis of Nasopharyngeal Carcinoma. Eur. J. Histochem. EJH 2021, 65, 3304.
- Zhang, X.; Wu, Y.; Sun, X.; Cui, Q.; Bai, X.; Dong, G.; Gao, Z.; Wang, Y.; Gao, C.; Sun, S. The PI3K/AKT/MTOR Signaling Pathway Is Aberrantly Activated in Primary Central Nervous System Lymphoma and Correlated with a Poor Prognosis. BMC Cancer 2022, 22, 1–11.
- Bhatti, M.; Ippolito, T.; Mavis, C.; Gu, J.; Cairo, M.S.; Lim, M.S.; Hernandez-Ilizaliturri, F.; Barth, M.J. Pre-Clinical Activity of Targeting the PI3K/Akt/MTOR Pathway in Burkitt Lymphoma. Oncotarget 2018, 9, 21820.
- Sindel, A.; McConnell, I.; Windle, J.; Sabo, R.; Chesney, A.; Lai, G.; Mauro, A.; Al-Juhaishi, T.; Rahmani, M.; Zweit, J. Role of the PI3K Pathway in the Pathogenesis of Marginal Zone Lymphoma. Blood 2018, 132, 4125.
- Okosun, J.; Wolfson, R.L.; Wang, J.; Araf, S.; Wilkins, L.; Castellano, B.M.; Escudero-Ibarz, L.; Al Seraihi, A.F.; Richter, J.; Bernhart, S.H. Recurrent MTORC1-Activating RRAGC Mutations in Follicular Lymphoma. Nat. Genet. 2016, 48, 183–188.
- Yu, D.; Zhang, Y.; Chen, G.; Xie, Y.; Xu, Z.; Chang, S.; Hu, L.; Li, B.; Bu, W.; Wang, Y. Targeting the PI3K/Akt/MTOR Signaling Pathway by Pterostilbene Attenuates Mantle Cell Lymphoma Progression. Acta Biochim. Biophys. Sin. 2018, 50, 782–792.

